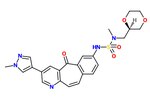
c-Met inhibitor
[4] c-Met is a receptor tyrosine kinase,[5] which can cause a wide variety of different cancers, such as renal, gastric and small cell lung carcinomas, central nervous system tumours, as well as several sarcomas[6] when its activity is dysregulated.
Targeting the ATP binding site of c-Met by small molecules inhibitors is one strategy for inhibition of the tyrosine kinase.
20 crystal structures with and without ligands have been published and in 2010 nearly a dozen small molecule c-Met inhibitors have been tested clinically.
[12] Receptor tyrosine kinases (RTKs) are a vital element in regulating many intracellular signal transduction pathways.
[11] c-Met dysregulation can be due to overexpression, gene amplification, mutation, a ligand-dependent auto- or paracrine loop or an untimely activation of RTK.
[13] Patients with aberrant c-Met activity usually have a poor prognosis, aggressive disease, increased metastasis and shortened survival.
The beta chain contains the intracellular tyrosine kinase domain and a tail on the C-terminal which is vital for the docking of substrates and downstream signalling.
[10] Phosphorylation occurs in tyrosines close to the C-terminus, creating a multi-functional docking site[10][18] which recruits adaptor proteins and leads to downstream signalling.
The signaling is mediated by Ras/Mapk, PI3K/Akt, c-Src and STAT3/5 and include cell proliferation, reduced apoptosis, altered cytoskeletal function and more.
The kinase domain usually consists of a bi-lobed structure, where the lobes are connected with a hinge region, adjacent to the very conserved ATP binding site.
[10] This was a key step in the progress of c-Met inhibitor development in that the acyl binding gives the terminal aryl group the ability to penetrate a deep hydrophobic pocket and so it enhances the potency of the compounds.
[19] AM7 and SU11274 offered the first proof that relatively selective c-Met inhibitors could be identified and that the inhibition leads to an anti-tumour effect in vivo.
[12] Even though the two classes are structurally different, they do share some properties: They both bind at the kinase hinge region (although they occupy different parts of the c-Met active site[20]) and they all aim to mimic the purine of ATP.
Structurally similar series of c-Met inhibitors in which a phenolic hinge binding element was linked to an arylamino-triazolopyridazine or aryl-triazolothiapyridazine.
Compounds with heterocyclic hinge binding elements (quinoline, pyridine, azaindole) linked to fused, nitrogen-dense heteroaromatics (triazolopyridazines, triazolopyrazines and triazolotriazines) have been described.
[12] JNJ-38877605, which contains a difluoro methyl linker and a bioavailable quinoline group, was undergoing clinical trials of Phase I for advanced and refractory solid tumours in 2010.
It led to slight regressions or stable disease in patients with papillary renal carcinoma and poorly differentiated gastric cancer.
[12] Tivantinib (ARQ197) is a selective, orally bioavailable,[17][21] clinically advanced low-molecular weight and well-tolerated c-MET inhibitor, which is currently[when?]
Tivantinib strongly inhibits c-Met autoactivation by selectively targeting the inactive form of the kinase between the N- and C- lobes and occupies the ATP binding site.
(e.g. XL184(Cabozantinib), XL880, ARQ197 ) [needs update] The use of c-Met inhibitors with other therapeutic agents could be crucial for overcoming potential resistance as well as for improving overall clinical benefit.