Glycogen storage disease type I
[7][8] Early research into GSD I identified numerous clinical manifestations falsely thought to be primary features of the genetic disorder.
However, after birth, the inability to maintain blood glucose from stored glycogen in the liver causes measurable hypoglycemia in no more than 1–2 hours after feedings.
Without proper dietary treatment after birth, prolonged hypoglycemia often leads to sudden lactic acidosis that can induce primary respiratory distress in the newborn period, as well as ketoacidosis.
[citation needed] In the early weeks of life, undiagnosed infants with GSD I tolerate persistent hypoglycemia and compensated lactic acidosis between feedings without symptoms.
After weeks to months without treatment with consistent oral carbohydrates, infants will progress to show clear symptoms of hypoglycemia and lactic acidosis.
Infants may present with paleness, clamminess, irritability, respiratory distress, and an inability to sleep through the night even in the second year of life.
When digestion of a meal is complete, insulin levels fall, and enzyme systems in the liver cells begin to remove glucose molecules from strands of glycogen in the form of G6P.
When fasting continues for more than a few hours, falling insulin levels permit catabolism of muscle protein and triglycerides from adipose tissue.
However, during and after an episode of low blood sugar, lactate levels will abruptly rise to exceed 15 mol/mL, the threshold for lactic acidosis.
Proper identification of lactic acidosis in undiagnosed children presents a challenge since the first symptoms are typically vomiting and dehydration, both of which mimic childhood infections like gastroenteritis or pneumonia.
This discomfort is an amplified form of the burning sensation a runner may feel in the quadriceps after sprinting, which is caused by a brief buildup of lactic acid.
It may also be caused by intracellular accumulation of glucose-6-phosphate with secondary shunting to pyruvate, which is converted into Acetyl-CoA, which is transported to the cytosol where the synthesis of fatty acids and cholesterol occurs.
Triglycerides above the 3.4 mmol/L (300 mg/dL) range may produce visible lipemia, and even a mild pseudohyponatremia due to a reduced aqueous fraction of the blood plasma.
Reductions in the mass of the liver are possible since most patients retain residual hepatic function that allows for the liberation of stored glycogen at a limited rate.
GSD Ib patients may present with splenomegaly, but this is connected to the use of filgrastim to treat neutropenia in this subtype, not comorbid hepatomegaly.
However, hepatic adenomas in GSD I uniquely involve diffuse Mallory hyaline deposition, which is otherwise commonly observed in focal nodular hyperplasia.
While the reason for the high prevalence of adenomas in GSD I is unclear, research since the 1970s has implicated serum glucagon as a potential driver.
In studies, patients who have been put on a dietary regimen to keep blood sugar in a normal range spanning 72 to 108 mg/dL (4.0 to 6.0 mmol/L) have shown a decreased likelihood of developing adenomas.
In many cases bone mineral density can increase and return to the normal range given proper metabolic control and calcium supplementation alone, reversing osteopenia.
In some cases, G-CSF formulated as pegfilgrastim, sold under the trade name Neulasta, may be used as a slow-acting alternative, requiring less frequent dosing.
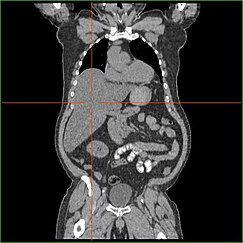
If hepatomegaly, fasting hypoglycemia, and poor growth are accompanied by lactic acidosis, hyperuricemia, hypertriglyceridemia, and enlarged kidneys by ultrasound, GSD I is the most likely diagnosis.
The differential diagnosis list includes glycogenoses types III and VI, fructose 1,6-bisphosphatase deficiency, and a few other conditions (page 5)[citation needed], but none are likely to produce all of the features of GSD I.
Administration of intramuscular or intravenous glucagon (0.25 to 1 mg, depending on age) or epinephrine produces little rise in blood sugar.
The diagnosis is definitively confirmed by liver biopsy with electron microscopy and assay of glucose-6-phosphatase activity in the tissue and/or specific gene testing, available in recent years.
To compensate for the inability of the liver to provide sugar, the total amount of dietary carbohydrates should approximate the 24-hour glucose production rate.
In the last 30 years, two methods have been used to achieve this goal in young children: (1) continuous nocturnal gastric infusion of glucose or starch; and (2) night-time feedings of uncooked cornstarch.
Sudden death from hypoglycemia has occurred due to malfunction or disconnection, and periodic cornstarch feedings are now preferred to continuous infusion.
Adenomas of the liver can develop in the second decade or later, with a small chance of later malignant transformation to hepatoma or hepatic carcinomas (detectable by alpha-fetoprotein screening).
Additional problems reported in adolescents and adults with GSD I have included hyperuricemic gout, pancreatitis, and chronic kidney failure.
With exceptions and qualifications, adult health and life span may also be fairly good, although the lack of effective treatment before the mid-1980s means information on long-term efficacy is limited.