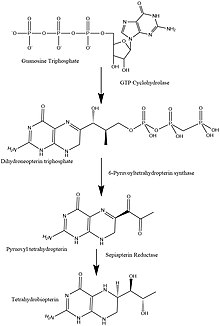
6-Pyruvoyltetrahydropterin synthase deficiency
Commonly reported symptoms are initial truncal hypotonia, subsequent appendicular hypertonia, bradykinesia, cogwheel rigidity, generalized dystonia, and marked diurnal fluctuation.
Other reported clinical features include difficulty in swallowing, oculogyric crises, somnolence, irritability, hyperthermia, and seizures.
Patients with mild phenotypes may deteriorate if given folate antagonists such as methotrexate, which can interfere with a salvage pathway through which dihydrobiopterin is converted into tetrahydrobiopterin via dihydrofolate reductase.
Treatment options include substitution with neurotransmitter precursors (levodopa, 5-hydroxytryptophan), monoamine oxidase inhibitors, and tetrahydrobiopterin.
To provide a basis for improving the understanding of the epidemiology, genotype–phenotype correlation and outcome of these diseases, their impact on the quality of life of patients, and for evaluating diagnostic and therapeutic strategies a patient registry was established by the noncommercial International Working Group on Neurotransmitter Related Disorders (iNTD).
PTPS deficiency can usually be detected by the same screening test used for PKU, because both disorders result in elevated levels of Phenylalanine.
PTPS deficiency occurs most commonly in Asian countries and this makes sense because of the incidence of this disease in Taiwanese Chinese being significantly higher than any other population.
Asbjørn Følling, a physician studying metabolic diseases, identified an excess of phenylpyruvate as the cause of a strange, musty odor from the urine of two Norwegian children.
The foundations for dietary restrictions were laid down by George Jervis and Host Bickel which is still one of the best ways to treat PKU.