Alpha-mannosidosis
[2] In humans it is known to be caused by an autosomal recessive genetic mutation in the gene MAN2B1, located on chromosome 19, affecting the production of the enzyme alpha-D-mannosidase, resulting in its deficiency.
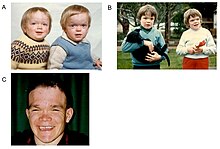
[1][6] This delays diagnosis, particularly as milder forms of the disease involve only mild to moderate intellectual disability, which progresses gradually during childhood or adolescence.
[2][9] Complete absence of functionality in this enzyme leads to death during early childhood due to deterioration of the central nervous system.
[9][8] Alpha-mannosidosis is a progressive disorder, and its presence should be suspected in patients with cognitive disabilities, skeletal changes (e.g., swollen joints, curved spine), hearing loss and recurrent infections.
Alpha-mannosidosis can impact a patient's quality of life in many ways, including their ability to live independently, socialise or find employment.
[2] Patients may present to doctors, nurses or health visitors at different stages of progression, and with different ad hoc symptoms, making the link to suspect a diagnosis of alpha-mannosidosis difficult.
[2] The condition is often diagnosed and treated using a multi-disciplinary approach, involving paediatricians, orthopaedics, ophthalmologists, otologists, neurologists, immunologists, neurosurgeons and physiotherapists.
After a full physical examination, physicians should focus on the known complications of alpha-mannosidosis, such as hydrocephalus, otitis media, hearing loss, dental caries, joint symptoms, kyphoscoliosis, and mental state.
[citation needed] Independent living will be difficult, and patients with alpha-mannosidosis may become socially isolated, and during the late stages of the disease they may become wheelchair-using, as they can no longer walk unaided.