Development and discovery of SSRI drugs
[5] Monoamine oxidase inhibitors (MAOIs) and tricyclic antidepressants (TCAs) were the first drugs to be developed for the treatment of depression, dating back to the early 1950s.
Because of their undesirable adverse-effect profile and high potential for toxicity, due to their non-selective pharmacological effects, strict regimens were needed for taking the drugs, which limited their use.
The SSRIs are the most significant class of antidepressants marketed in recent years and are one of the major medicinal discoveries of the last few decades.
SSRIs were the first drugs to establish a theoretical pathophysiological role for 5-HT in affective illnesses and in the broad spectrum of anxiety disorders.
The work which eventually led to the discovery of fluoxetine began at Eli Lilly and Company in 1970 as a collaboration between Bryan Molloy and Ray Fuller.
Molloy and fellow Eli Lilly chemist Klaus Schmiegel synthesized a series of dozens of its derivatives.
[10] This test showed the compound later named fluoxetine to be the most potent and selective inhibitor of serotonin reuptake of the series.
[13] Introduction of fluoxetine to the market is hailed as a miracle drug for the treatment of depression because it had fewer adverse effects, simpler dosing strategies and greater margin of safety when overdoses were consumed and thus it had better adherence, compared to the older antidepressants (TCAs and MAOIs).
[6] Since then the number of drugs in the SSRI class has become bigger and there are now six (fluoxetine, paroxetine, citalopram, escitalopram, sertraline, and fluvoxamine),[5][9] as demonstrated in table 1.
Precise mechanism of antidepressant activity of SSRIs remains somewhat uncertain, but a number of biochemical functions associated with SSRI treatment have been established.
This delay is caused by the time it takes 5-HT to downregulate 5-HT1A autoreceptors and turn on the neuro impulse flow of the 5-HT neuron.
Table 3 Comparative pharmacology of SSRIs tmax = Time to peak plasma level after oral dose; VD = Volume of distribution; t1/2 = Elimination half-life It is recognized that both the position and the type of substitution on an aromatic moiety of the SSRI compounds are important for the higher specificity to SERT.
[38] SSRI's are by definition selective, but they also bind to the homologous NET and DAT, although with much lower affinity than to their principal target SERT.
The selectivity of SSRIs for SERT is notable in that only one or two different functional group substituents are sufficient to convert an SSRI into a norepinephrine reuptake inhibitor (NRI) with higher affinity to NE.
However, despite the sharing of the same mechanism of action, SSRIs differ in their potency and selectivity in inhibiting 5-HT re-uptake and many of them have important effects on other transporters and receptors.
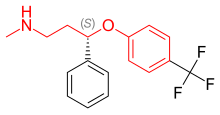
[6] Table 2 Comparison of the chemical properties of SSRI drugs Compounds containing an aryloxypropylamine motif in their structure, demonstrated in figure 3a, are known as monoamine reuptake inhibitors.
Drugs containing this privileged structural motif, where R1 and R2 are aryls or heteroaryls, preferable phenyl, possess a selectivity profile for NET and SERT.
The advantage of being 5-HT2C antagonist is that it has a stimulatory effect and many patients have experienced an increase in energy, concentration and focus and a decrease in fatigue from the very first dose.
As demonstrated in table 2, paroxetine also inhibits the NOSs enzyme which could be the reason for its sexual dysfunction adverse effect, especially in men.
[18] Paroxetine shows the highest affinity for muscarinic receptors of all the SSRIs which results in weak anticholinergic activity and therefore undesirable adverse effects.
[44] While scientists were trying to create a new antidepressant to inhibit the NE re-uptake they accidentally synthesised two new compounds, named talopram and talsupram.
The two compounds where not marketed in spite of being potent SNRIs because a number of suicide attempts were reported in clinical trials.
[6][18] Tametraline, a compound synthesized in 1978 by Pfizer, was shown to be a potent NE and DA re-uptake inhibitor with animal studies.
SERT contains approximately 630 amino acids that are predicted to form 12 transmembrane alpha-helixes (TMs) which are connected with intra- and extracellular loops (ILs and ELs).
Therefore, the crystal structure of LeuT and its transport mechanism have been proven to be a good model system for the study of NSS proteins.
Because of the (S)-enantiomers opposite chirality to the (R)-enantiomer the rest of the molecule is reversed in the HBP, where the amine tail points towards the extracellular space and interacts with the N-terminal of Leu400, Asp401 and Ala319 (amino acids which are a part of the TM10).
The tetralin (tetrahydronaphthalene) on the other end of sertralines structure is in contact with Leu400, Asp401 and Thr409 (which are a part of the TM10) as well as the molecule interacts with Ala319 of the EL4 hairpin loop and Arg30 and Gin34 of the TM1, where the amine tail points towards the cytoplasm.
The bound sertraline molecule has its dichlorophenyl ring rotated about the C4-C13 bond by 180 degrees compared to the free drug.
[38] Andersen et al. were able to generate a model of the (S)-citalopram binding site in human SERT by combining mutational analysis and comparative modeling where they found out that Asn-177 and Phe-341 where key determinants for (S)-citalopram potency and high affinity inhibition[47] in addition to Tyr-95, Asp-98, Ile-172 and Ser438 previously described, where three functional groups of the inhibitors structure bind to the transporters amino acids.