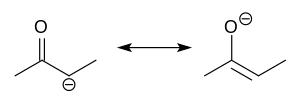
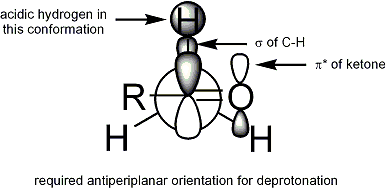
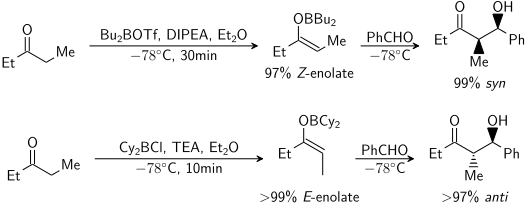
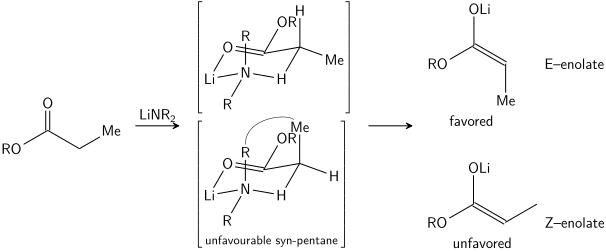
Enolate
A weaker base such as an alkoxide, which reversibly deprotonates the substrate, affords the more thermodynamically stable benzylic enolate.
In most cases, it is not known which, if any, intermediates are monomeric or oligomeric in nature; nonetheless, the Ireland model remains a useful tool for understanding enolates.
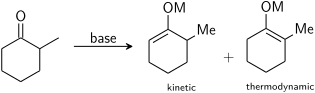
In general, kinetic enolates are favored by cold temperatures, conditions that give relatively ionic metal–oxygen bonding, and rapid deprotonation using a slight excess of a strong, sterically hindered base.
Thermodynamic enolates are favored by longer equilibration times at higher temperatures, conditions that give relatively covalent metal–oxygen bonding, and use of a slight sub-stoichiometric amount of strong base.
This provides one of the best understood synthetic strategies to introduce chemical complexity in natural product and total syntheses.
The selectivity is determined by both the steric and electronic effects on the α-carbons as well as the precise base used (see figure ""Masked functionality" for regiospecific enolate formation" for an example of this).
Enolate formation will be thermodynamically favoured at the most acidic proton which depends on the electronic stabilization of the resulting anion.
However, the selectivity can be reversed by sterically hindering the thermodynamic product and therefore kinetically favouring deprotonation at the other α-carbon centre.
This process is first described by Gilbert Stork[22] who is best known for his contributions to the study of selective enolate formation methods in organic synthesis.
The "masked functionality" approach to regiospecific enolate formation has been widely used in the total synthesis of natural products.
For example, in the total synthesis of the steroid hormone progesterone,[23] Stork and co-workers used the "masked functionality" to stereospecifically construct one of the quaternary carbons in the molecule.
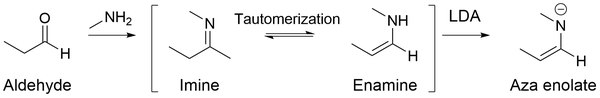
The major benefit of using aza enolates is that they don't undergo self-condensation (i.e. aldol reaction for aldehydes) in a basic or neutral solution, but rather they favor alkylation on the alpha-carbon.
[25] Through nitrogen lone pair conjugation, β-carbon becomes a nucleophilic site, permitting aza enolates to undergo alkylation reactions.
[27] Thus, aza enolates can react with numerous electrophiles like epoxides and alkyl halides to form a new carbon-carbon bond on β-carbon.
To counter this effect, the nucleophilic aza enolates easily react with epoxides to reduce their ring strains.
Aza enolates can also be formed with Grignard reagents and react with other soft electrophiles, including Michael receptors.