Huntington's disease
[2] In the early stages, subtle personality changes, problems in cognition and physical skills, irritability, and mood swings occur, all of which may go unnoticed,[21][22] and these usually precede the motor symptoms.
Early damage is most evident in the subcortical basal ganglia, initially in the striatum, but as the disease progresses, other areas of the brain are also affected, including regions of the cerebral cortex.
It also acts as an antiapoptotic agent preventing programmed cell death and controls the production of brain-derived neurotrophic factor, a protein that protects neurons and regulates their creation during neurogenesis.
[54] Initially, damage to the brain is regionally specific with the dorsal striatum in the subcortical basal ganglia being primarily affected, followed later by cortical involvement in all areas.
[59] Because of the basal ganglia's inability to inhibit movements, individuals affected by it inevitably experience a reduced ability to produce speech and swallow foods and liquids (dysphagia).
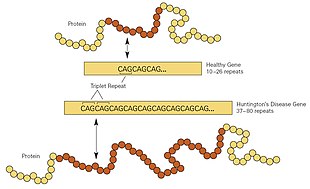
Even before the onset of symptoms, genetic testing can confirm if an individual or embryo carries an expanded copy of the trinucleotide repeat (CAG) in the HTT gene that causes the disease.
[26] Other factors taken into account when considering testing include the possibility of discrimination and the implications of a positive result, which usually means a parent has an affected gene and that the individual's siblings will be at risk of inheriting it.
Some forms of preimplantation genetic diagnosis—non-disclosure or exclusion testing—allow at-risk people to have HD-free offspring without revealing their own parental genotype, giving no information about whether they themselves are destined to develop HD.
[76][77] Obtaining a prenatal diagnosis for an embryo or fetus in the womb is also possible, using fetal genetic material acquired through chorionic villus sampling.
[26] Weight loss and problems in eating due to dysphagia and other muscle discoordination are common, making nutrition management increasingly important as the disease advances.
A recent study showed that the stromal processing peptidase (SPP), a synthetic enzyme found in plant chloroplasts, prevented the aggregation of proteins associated with Huntington's disease.
Genetic counseling benefits these individuals by updating their knowledge, seeking to dispel any unfounded beliefs that they may have, and helping them consider their future options and plans.
[26] The worldwide prevalence of HD is 5–10 cases per 100,000 persons,[102][103] but varies greatly geographically as a result of ethnicity, local migration and past immigration patterns.
[114][113] Independently of Gorman and Waters, both students of Dunglison at Jefferson Medical College in Philadelphia,[115] Johan Christian Lund [no] (1830–1906) also produced an early description in 1860.
[113] He specifically noted that in Setesdalen, a secluded mountain valley in Norway, the high prevalence of dementia was associated with a pattern of jerking movement disorders that ran in families.
Examining the combined medical history of several generations of a family exhibiting similar symptoms, he realized their conditions must be linked; he presented his detailed and accurate definition of the disease as his first paper.
[111][117]Sir William Osler was interested in the disorder and chorea in general, and was impressed with Huntington's paper, stating, "In the history of medicine, there are few instances in which a disease has been more accurately, more graphically or more briefly described.
"[118][113][119] Osler's continued interest in HD, combined with his influence in the field of medicine, helped to rapidly spread awareness and knowledge of the disorder throughout the medical community.
[120][115] The strong inheritance pattern prompted several researchers, including Smith Ely Jelliffe, to attempt to trace and connect family members of previous studies.
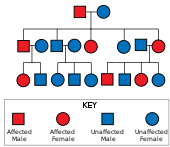
[121] Jelliffe's research roused the interest of his college friend, Charles Davenport, who commissioned Elizabeth Muncey to produce the first field study on the East Coast of the United States of families with HD and to construct their pedigrees.
Researchers have found contrary evidence; for instance, the community of the family studied by George Huntington openly accommodated those who exhibited symptoms of HD.
[127] In the same time, key discoveries concerning the mechanisms of the disorder were being made, including the findings by Anita Harding's research group on the effects of the gene's length.
There was controversy when Charles Davenport proposed in 1910 that compulsory sterilization and immigration control be used for people with certain diseases, including HD, as part of the eugenics movement.
[139][140] As with other untreatable genetic conditions with a later onset, it is ethically questionable to perform presymptomatic testing on a child or adolescent since there would be no medical benefit for that individual.
[143] In 1968, after experiencing HD in his wife's family, Dr. Milton Wexler was inspired to start the Hereditary Disease Foundation (HDF), with the aim of curing genetic illnesses by coordinating and supporting research.
[153] The safety of RNA interference, and allele-specific oligonucleotide (ASO) methods of gene silencing has been demonstrated in mice and the larger primate macaque brain.
[156] The first gene silencing trial involving humans with HD began in 2015, testing the safety of IONIS-HTTRx, produced by Ionis Pharmaceuticals and led by UCL Institute of Neurology.
[157][158] Mutant huntingtin was detected and quantified for the first time in cerebrospinal fluid from Huntington's disease mutation-carriers in 2015 using a novel "single-molecule counting" immunoassay,[159] providing a direct way to assess whether huntingtin-lowering treatments are achieving the desired effect.
[160][161] A phase 3 trial of this compound, renamed tominersen and sponsored by Roche Pharmaceuticals, began in 2019 but was halted in 2021 after the safety monitoring board concluded that the risk-benefit balance was unfavourable.
[171] Compounds trialled that have failed to prevent or slow the progression of Huntington's disease include remacemide, coenzyme Q10, riluzole, creatine, minocycline, ethyl-EPA, phenylbutyrate and dimebon.