ALS
[20] In 1869, the connection between the symptoms and the underlying neurological problems was first described by French neurologist Jean-Martin Charcot, who in 1874 began using the term amyotrophic lateral sclerosis.
Each individual diagnosed with the condition will sit at a unique place at the intersection of these complex and overlapping subtypes, which presents a challenge to diagnosis, understanding, and prognosis.
[6] A rarer type of classical ALS affecting around 3% of patients is respiratory-onset,[9] in which the initial symptoms are difficulty breathing (dyspnea) upon exertion, at rest, or while lying flat (orthopnea).
[25] Primary lateral sclerosis (PLS) is a subtype of the overall ALS category which accounts for about 5% of all cases and only affects the upper motor neurons in the arms, legs, and bulbar region.
[26] Progressive muscular atrophy (PMA) is another subtype that accounts for about 5% of the overall ALS category and affects lower motor neurons in the arms, legs, and bulbar region.
[26] While PMA is associated with longer survival on average than classical ALS, it is still progressive over time, eventually leading to respiratory failure and death.
Sensory nerves and the autonomic nervous system are generally unaffected, meaning the majority of people with ALS maintain hearing, sight, touch, smell, and taste.
If the arms are affected first, they may experience difficulty with tasks requiring manual dexterity, such as buttoning a shirt, writing, or turning a key in a lock.
[41] Brief periods of stabilization ("plateaus") and even small reversals in ALSFRS-R score are not uncommon, due to the fact the tool is subjective, can be affected by medication, and different forms of compensation for changes in function.
[44] In later stages of the disorder, aspiration pneumonia can develop, and maintaining a healthy weight can become a significant problem that may require the insertion of a feeding tube.
[44] As the diaphragm and intercostal muscles of the rib cage that support breathing weaken, measures of lung function such as vital capacity and inspiratory pressure diminish.
[50] Access to palliative care is recommended from an early stage to explore options, ensure psychosocial support for the patient and caregivers, and to discuss advance healthcare directives.
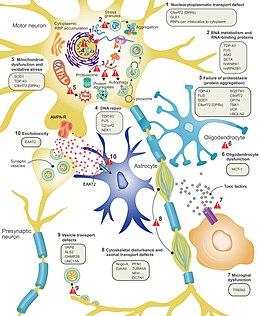
A multi-step liability threshold model for ALS proposes that cellular damage accumulates over time due to genetic factors present at birth and exposure to environmental risks throughout life.
[9] The genes known to be involved in ALS can be grouped into three general categories based on their normal function: protein degradation, the cytoskeleton, and RNA processing.
[88] Excitotoxicity, or nerve cell death caused by high levels of intracellular calcium due to excessive stimulation by the excitatory neurotransmitter glutamate, is a mechanism thought to be common to all forms of ALS.
Riluzole, a drug that modestly prolongs survival in ALS, inhibits glutamate release from pre-synaptic neurons; however, it is unclear if this mechanism is responsible for its therapeutic effect.
While a magnetic resonance imaging (MRI) is often normal in people with early-stage ALS, it can reveal evidence of other problems that may be causing the symptoms, such as a spinal cord tumor, multiple sclerosis, a herniated disc in the neck, syringomyelia, or cervical spondylosis.
[91] Because the prognosis of ALS and closely related subtypes of motor neuron disease are generally poor, neurologists may carry out investigations to evaluate and exclude other diagnostic possibilities.
Disorders of the neuromuscular junction, such as myasthenia gravis (MG) and Lambert–Eaton myasthenic syndrome, may also mimic ALS, although this rarely presents diagnostic difficulty over time.
Nonetheless, the absence of other neurological features that develop inexorably with ALS means that, over time, the distinction will not present any difficulty to the experienced neurologist; where doubt remains, EMG may be helpful.
[5] The most commonly used measurement is upright forced vital capacity (FVC), but it is a poor detector of early respiratory failure and is not a good choice for those with bulbar symptoms, as they have difficulty maintaining a tight seal around the mouthpiece.
[96] Sniff nasal inspiratory pressure (SNIP) is a rapid, convenient test of diaphragm strength that is not affected by bulbar muscle weakness.
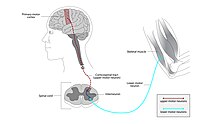
The person with ALS will continue to lose motor function, making communication increasingly difficult and sometimes leading to locked-in syndrome, in which they are completely paralyzed except for their eye muscles.
[101] Gentle, low-impact aerobic exercise such as performing activities of daily living, walking, swimming, and stationary bicycling can strengthen unaffected muscles, improve cardiovascular health, and help people fight fatigue and depression.
Occupational therapists can provide or recommend equipment and adaptations to enable ALS people to retain as much safety and independence in activities of daily living as possible.
[5] Palliative care, which relieves symptoms and improves the quality of life without treating the underlying disease, should begin shortly after someone is diagnosed with ALS.
[107] Late in the disease course, difficulty speaking due to muscle weakness (dysarthria) and cognitive dysfunction may impair their ability to communicate their wishes regarding care.
If people with ALS or their family members are reluctant to discuss end-of-life issues, it may be useful to use the introduction of gastrostomy or noninvasive ventilation as an opportunity to bring up the subject.
[20] In 1850, François-Amilcar Aran was the first to describe a disorder he named "progressive muscular atrophy", a form of ALS in which only the lower motor neurons are affected.
[129] In 1869, the connection between the symptoms and the underlying neurological problems was first described by Jean-Martin Charcot, who initially introduced the term amyotrophic lateral sclerosis in his 1874 paper.