Molecular modelling
Molecular models typically describe atoms (nucleus and electrons collectively) as point charges with an associated mass.
This function, referred to as a potential function, computes the molecular potential energy as a sum of energy terms that describe the deviation of bond lengths, bond angles and torsion angles away from equilibrium values, plus terms for non-bonded pairs of atoms describing van der Waals and electrostatic interactions.
[2] The common force fields in use today have been developed by using chemical theory, experimental reference data, and high level quantum calculations.
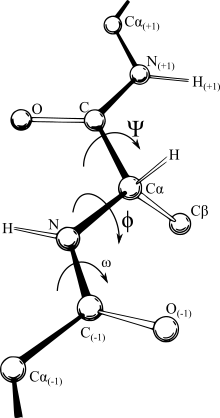
Yet the comparatively rigid nature of bonds which occur between specific atoms, and in essence, defines what is meant by the designation molecule, make an internal coordinate system the most logical representation.
Unfortunately, continuous motions in Cartesian space often require discontinuous angular branches in internal coordinates, making it relatively hard to work with force fields in the internal coordinate representation, and conversely a simple displacement of an atom in Cartesian space may not be a straight line trajectory due to the prohibitions of the interconnected bonds.
This can dominate the calculation time of the potential itself and in long chain molecules introduce cumulative numerical inaccuracy.
[3] Currently, the fastest and most accurate torsion to Cartesian conversion is the Natural Extension Reference Frame (NERF) method.
[3] Molecular modelling methods are used routinely to investigate the structure, dynamics, surface properties, and thermodynamics of inorganic, biological, and polymeric systems.