Neutral network (evolution)
[29] Kimura computed the rate of nucleotide substitutions in a population (i.e. the average time for one base pair replacement to occur within a genome) and found it to be ~1.8 years.
[31] DNA is found, predominantly, as fully base paired double helices, while biological RNA is single stranded and often exhibits complex base-pairing interactions.
These are due to its increased ability to form hydrogen bonds, a fact which stems from the existence of the extra hydroxyl group in the ribose sugar.
[32] Waterman gave the first graph theoretic description of RNA secondary structures and their associated properties, and used them to produce an efficient minimum free energy (MFE) folding algorithm.
[33] An RNA secondary structure can be viewed as a diagram over N labeled vertices with its Watson-Crick base pairs represented as non-crossing arcs in the upper half plane.
[37] Later, Waterman and Temple (1986) produced a polynomial time dynamic programming (DP) algorithm for predicting general RNA secondary structure.
[38] while in the year 1990, John McCaskill presented a polynomial time DP algorithm for computing the full equilibrium partition function of an RNA secondary structure.
M. Zuker, implemented algorithms for computation of MFE RNA secondary structures[40] based on the work of Nussinov et al.,[35] Smith and Waterman[34] and Studnicka, et al.[41] Later L. Hofacker (et al., 1994),[42] presented The Vienna RNA package, a software package that integrated MFE folding and the computation of the partition function as well as base pairing probabilities.
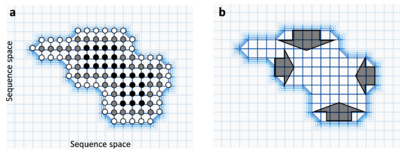
These two facts lead them to conclude that the sequence space seemed to be percolated by neutral networks of nearest neighbor mutants that fold to the same structure.
They showed that the networks are connected and percolate sequence space if the fraction of neutral nearest neighbors exceeds λ*, a threshold value.
Key results of this analysis where concerned with threshold functions for density and connectivity for neutral networks as well as Schuster's shape space conjecture.