Organolithium reagent
Studies of organolithium reagents began in the 1930s and were pioneered by Karl Ziegler, Georg Wittig, and Henry Gilman.
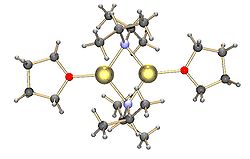
[6][12] In aryllithium complexes, the lithium cation coordinates to a single carbanion center through a Li−C σ type bond.
[7][14] Formation of aggregates is influenced by electrostatic interactions, the coordination between lithium and surrounding solvent molecules or polar additives, and steric effects.
[7] A basic building block toward constructing more complex structures is a carbanionic center interacting with a Li3 triangle in an η3- fashion.
Methyllithium exists as tetramers in a cubane-type cluster in the solid state, with four lithium centers forming a tetrahedron.
[7] This difference can arise from the method of preparation of silyllithiums, the steric hindrance caused by the bulky alkyl substituents on silicon, and the less polarized nature of Si−Li bonds.
[20] Organolithium compounds bind Lewis bases such as tetrahydrofuran (THF), diethyl ether (Et2O), tetramethylethylene diamine (TMEDA) or hexamethylphosphoramide (HMPA).
[7][22] One question surrounding the structure-reactivity relationship is whether there exists a correlation between the degree of aggregation and the reactivity of organolithium reagents.
[25] A series of solution kinetics studies of LDA-mediated reactions suggest that lower aggregates of enolates do not necessarily lead to higher reactivity.
[7] Toward alkyllithium reagents, TMEDA functions as a donor ligand, reduces the degree of aggregation,[5] and increases the nucleophilicity of these species.
Some of the most common applications of organolithium reagents in synthesis include their use as nucleophiles, strong bases for deprotonation, initiator for polymerization, and starting material for the preparation of other organometallic compounds.
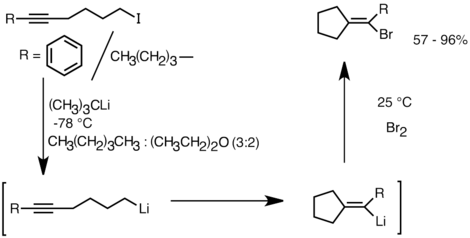
First, it is possible for the product cyclic organolithium species to react with electrophiles, whereas it is often difficult to trap a radical intermediate of the corresponding structure.
The limitations of intramolecular carbolithiation include difficulty of forming 3 or 4-membered rings, as the intermediate cyclic organolithium species often tend to undergo ring-openings.
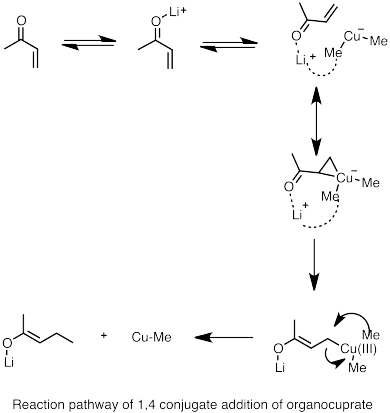
Secondly, adding donor ligands to the reaction forms heteroatom-stabilized lithium species which favors 1,4 conjugate addition.
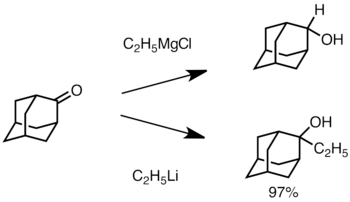
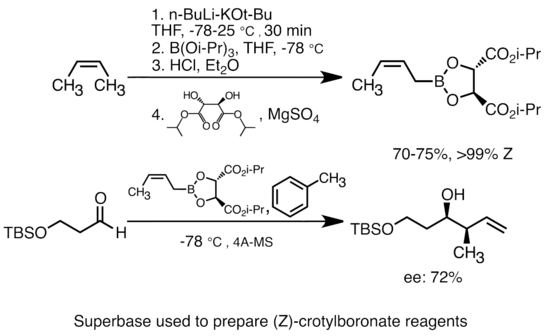
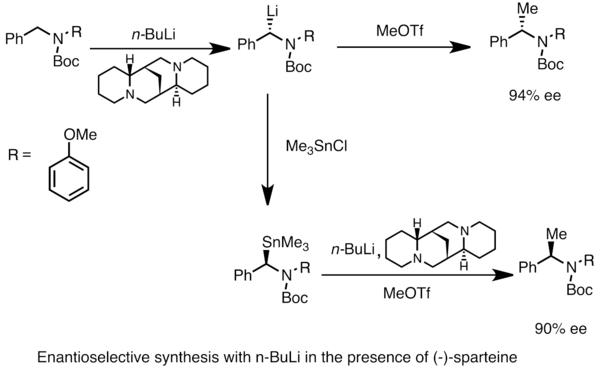
[43][44] Organolithium reagents can also perform enantioselective nucleophilic addition to carbonyl and its derivatives, often in the presence of chiral ligands.
[6] HMPA has been shown to increase reaction rate and product yields, and the reactivity of aryllithium reagents is often enhanced by the addition of potassium alkoxides.
Lithiation often occurs at a position α to electron withdrawing groups, since they are good at stabilizing the electron-density of the anion.
Metalation of allyl ether with alkyllithium or LDA forms an anion α to the oxygen, and can proceed to 2,3-Wittig rearrangement.
Addition of donor ligands such as TMEDA and HMPA can increase metalation rate and broaden substrate scope.
This reaction proceeds through deprotonation by organolithium reagents at the positions α to the direct metalation group (DMG) on the aromatic ring.
This generates a complex-induced proximity effect, which directs deprotonation at the α position to form an aryllithium species that can further react with electrophiles.
In the presence of two DMGs, metalation often occurs ortho to the stronger directing group, though mixed products are also observed.
Lithium enolate formation can be generalized as an acid–base reaction, in which the relatively acidic proton α to the carbonyl group (pK =20-28 in DMSO) reacts with organolithium base.
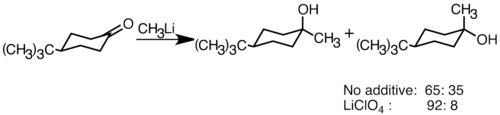
Many factors influence the outcome of enolate stereochemistry, such as steric effects, solvent, polar additives, and types of organolithium bases.
[55] In this assumption, a monomeric LDA reacts with the carbonyl substrate and form a cyclic Zimmerman–Traxler type transition state.
Organocopper, organotin, organosilicon, organoboron, organophosphorus, organocerium and organosulfur compounds are frequently prepared by reacting organolithium reagents with appropriate electrophiles.
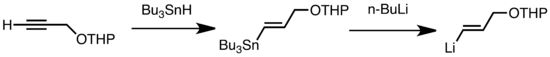
[47] The advantage of Li/Sn exchange is that the tri-alkylstannane precursors undergo few side reactions, as the resulting n-Bu3Sn byproducts are unreactive toward alkyllithium reagents.
[48] Industrial preparation of organolithium reagents is achieved using this method by treating the alkyl chloride with metal lithium containing 0.5–2% sodium.
This is the most common method for preparing alkynyllithium reagents, because the terminal hydrogen bound to the sp carbon is very acidic and easily deprotonated.
In the Shapiro reaction, two equivalents of strong alkyllithium base react with p-tosylhydrazone compounds to produce the vinyllithium, or upon quenching, the olefin product.