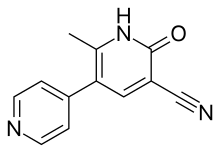
PDE3 inhibitor
[2] Well controlled studies have shown that these drugs generally increase mortality,[3] when used for the therapy of acute heart failure, so they have to be applied under close observation.
[1] The most important adverse effects when used for the therapy of acute heart failure are arrhythmia, thrombocytopenia and increased transaminase levels.
Inhibition of the PDE isoenzyme 3 leads to an increase of intracellular concentrations of the second messenger cyclic adenosine monophosphate (cAMP).
Most studies used analogues of the nucleotide substrates or derivatives of natural product inhibitors such as xanthine (e.g. theophylline) and papaverine.
These atoms are believed to mimic the electrophilic center in the phosphate group in cAMP and are confirmed as the primary site of binding.
Alkyl groups, limited to either methyl or ethyl, on the heterocyclic ring usually enhance potency, with occasional exceptions.
There is general agreement about this inhibitor potency: lactam ≥ alkyl-CONH- ≥ imidazoyl = pyridine in place of the central phenyl with its nitrogen in the analogous 4 position ≥ alkyl-S- > simple ether > halide = amine > imidazolium (which is totally inactive).
[6] Identification of features common to the most selective inhibitors has led to a "five-point model" with: Theophylline is a non-selective agent.