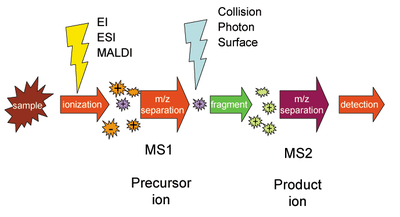
Tandem mass spectrometry
[citation needed] When tandem MS is performed with an in space design, the instrument must operate in one of a variety of modes.
Similar to the precursor-ion scan, this technique is also useful in the selective identification of closely related class of compounds in a mixture.
If the product ions persist in their non-equilibrium state for a moderate amount of time before auto-dissociation this process is called metastable fragmentation.
[16] EISA enables fragmentation data acquisition on MS1 mass analyzers such as time-of-flight and single quadrupole instruments.
[26][27] Freely available large scale high resolution tandem mass spectrometry databases exist (e.g. METLIN with 850,000 molecular standards each with experimental CID MS/MS data),[28] and are typically used to facilitate small molecule identification.
[31][32] Similar to electron-capture dissociation, ETD induces fragmentation of cations (e.g. peptides or proteins) by transferring electrons to them.
It was invented by Donald F. Hunt, Joshua Coon, John E. P. Syka and Jarrod Marto at the University of Virginia.
[34] ETD cleaves randomly along the peptide backbone (c and z ions) while side chains and modifications such as phosphorylation are left intact.
The technique only works well for higher charge state ions (z>2), however relative to collision-induced dissociation (CID), ETD is advantageous for the fragmentation of longer peptides or even entire proteins.
[35] Electron-transfer and higher-energy collision dissociation (EThcD) is a combination ETD and HCD where the peptide precursor is initially subjected to an ion/ion reaction with fluoranthene anions in a linear ion trap, which generates c- and z-ions.
[23] This method employs dual fragmentation to generate ion- and thus data-rich MS/MS spectra for peptide sequencing and PTM localization.
This process is called infrared multiphoton dissociation (IRMPD) and is often accomplished with a carbon dioxide laser and an ion trapping mass spectrometer such as a FTMS.
[44] In the BIRD method, the entire mass spectrometer vacuum chamber is heated to create infrared light.
BIRD uses this radiation to excite increasingly more energetic vibrations of the ions, until a bond breaks, creating fragments.
[citation needed] With surface-induced dissociation (SID), the fragmentation is a result of the collision of an ion with a surface under high vacuum.
Additionally, these surfaces are composed of rigid fluorocarbon chains, which don't significantly dampen the energy of the projectile ions.
The fluorocarbon chains are also beneficial because of their ability to resist facile electron transfer from the metal surface to the incoming ions.
[48] SID's ability to produce subcomplexes that remain stable and provide valuable information on connectivity is unmatched by any other dissociation technique.
Three different methods for this technique include analyzing the characterization of topology, intersubunit connectivity, and the degree of unfolding for protein structure.
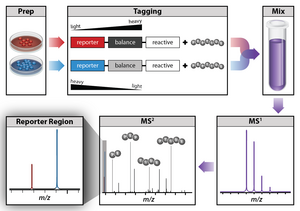
Isobaric tag labeling enables simultaneous identification and quantification of proteins from multiple samples in a single analysis.
[citation needed] An isobaric tag for relative and absolute quantitation (iTRAQ) is a reagent for tandem mass spectrometry that is used to determine the amount of proteins from different sources in a single experiment.
[57][58][59] It uses stable isotope labeled molecules that can form a covalent bond with the N-terminus and side chain amines of proteins.
The iTRAQ reagents are used to label peptides from different samples that are pooled and analyzed by liquid chromatography and tandem mass spectrometry.
The fragmentation of the attached tag generates a low molecular mass reporter ion that can be used to relatively quantify the peptides and the proteins from which they originated.
TMT reagents can be used to simultaneously analyze 2 to 11 different peptide samples prepared from cells, tissues or biological fluids.
The progress in data independent acquisition (DIA) enabled multiplexed quantitative proteomics with non-isobaric mass tags and a new method called plexDIA introduced in 2021.
[63] This new approach increases the number of data points by parallelizing both samples and peptides, thus achieving multiplicative gains.
[83][84] The development of tandem mass spectrometry screening in the early 1990s led to a large expansion of potentially detectable congenital metabolic diseases that affect blood levels of organic acids.
[86] Tandem mass spectrometry cannot be applied for single-cell analyses as it is insensitive to analyze such small amounts of a cell.
[87] Tandem mass spectrometry will be a useful tool for protein characterization, nucleoprotein complexes, and other biological structures.