Third-generation sequencing
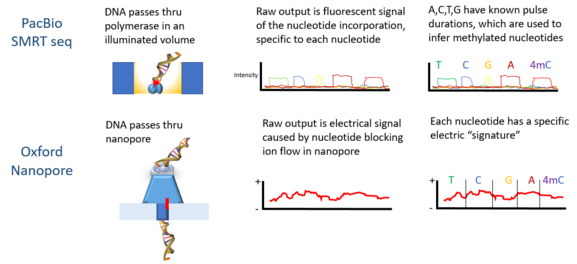
Signals are in the form of fluorescent light emission from each nucleotide incorporated by a DNA polymerase bound to the bottom of the zL well.
Stratos Genomics spaces out the DNA bases with polymeric inserts, "Xpandomers", to circumvent the signal to noise challenge of nanopore ssDNA reading.
It is expected that these longer read lengths will alleviate numerous computational challenges surrounding genome assembly, transcript reconstruction, and metagenomics among other important areas of modern biology and medicine.
The current generation of sequencing technologies rely on laboratory techniques such as ChIP-sequencing for the detection of epigenetic markers.
Third generation sequencing may enable direct detection of these markers due to their distinctive signal from the other four nucleotide bases.
[6] Since minimal sample preprocessing is required in comparison to second generation sequencing, smaller equipments could be designed.
This sequencing machine is roughly the size of a regular USB flash drive and can be used readily by connecting to a laptop.
These advantages of third generation sequencing may be well-suited in hospital settings where quick and on-site data collection and analysis is demanded.
The high error rates involved with third generation sequencing are inevitably problematic for the purpose of characterizing individual differences that exist between members of the same species.
Such reference based assembly is quick and easy but has the disadvantage of “hiding" novel sequences and large copy number variants.
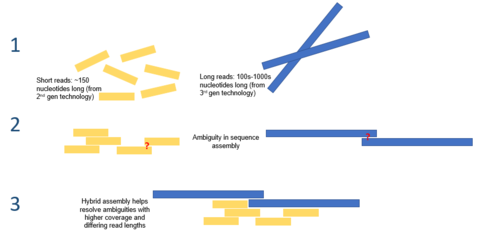
Given the short reads produced by the current generation of sequencing technologies, de novo assembly is a major computational problem.
[7] Long read lengths offered by third generation sequencing may alleviate many of the challenges currently faced by de novo genome assemblies.
DNA modifications and resulting gene expression can vary across cell types, temporal development, with genetic ancestry, can change due to environmental stimuli and are heritable.
The current most common methods for examining methylation state require an assay that fragments DNA before standard second generation sequencing on the Illumina platform.
[12] Here they found that in E. coli, which has a known methylome, event windows of 5 base pairs long can be used to divide and statistically analyze the raw MinION electrical signals.
[15] Other forms of DNA modifications – from heavy metals, oxidation, or UV damage – are also possible avenues of research using Oxford Nanopore and PacBio third generation sequencing.
MinION has low throughput; since multiple overlapping reads are hard to obtain, this further leads to accuracy problems of downstream DNA modification detection.
Both the hidden Markov model and statistical methods used with MinION raw data require repeated observations of DNA modifications for detection, meaning that individual modified nucleotides need to be consistently present in multiple copies of the genome, e.g. in multiple cells or plasmids in the sample.
While expression levels can be more or less accurately depicted by second generation sequencing (we can assume that actual abundances of the population of transcripts are randomly sampled), transcript-level information still remains an important challenge.
Alternative splicing (AS) is the process by which a single gene may give rise to multiple distinct mRNA transcripts and consequently different protein translations.
[16] Its evidence suggested that existing methods are generally weak in assembling transcripts, though the ability to detect individual exons are relatively intact.
While error rates remain high, third generation sequencing technologies have the capability to produce much longer read lengths.
[21] Pacific Bioscience has introduced the iso-seq platform, proposing to sequence mRNA molecules at their full lengths.
The relatively long reads allowed for sequencing of a near-complete viral genome to high accuracy (97–99% identity) directly from a primary clinical sample.
[24][25] In this context the PacBio error rate was comparable to that of shorter reads from 454 and Illumina's MiSeq sequencing platforms.
[citation needed] MinION's high error rate (~10-40%) prevented identification of antimicrobial resistance markers, for which single nucleotide resolution is necessary.
[23] Ease of carryover contamination when re-using the same flow cell (standard wash protocols don’t work) is also a concern.
Furthermore, performing accurate species identification for bacteria, fungi and parasites is very difficult, as they share a larger portion of the genome, and some only differ by <5%.