Argininosuccinate lyase
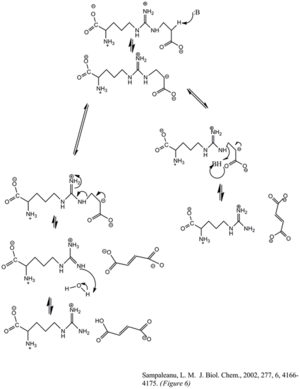
The enzyme argininosuccinate lyase (EC 4.3.2.1, ASL, argininosuccinase; systematic name 2-(N ω-L-arginino)succinate arginine-lyase (fumarate-forming)) catalyzes the reversible breakdown of argininosuccinate: Located in liver cytosol, it is the fourth enzyme of the urea cycle and involved in the biosynthesis of arginine in all species and the production of urea in ureotelic species.
ASL disorder is associated with considerable clinical and genetic heterogeneity which is considered to reflect the extensive intragenic complementation occurring among individual patients.
Although there is no consensus of the catalytic acid that donates the proton to the imine functional group of the arginine product, some mutagenesis studies show serine 283 may be involved.
ASL catalyzes the fourth step in the cycle, following the action of argininosuccinate synthetase (ASS) in the liver cytosol.
[4] Within the superfamily, ASL is most closely related to δ-crystallin in amino acid sequence and in protein fold structure.
The similarities have led researches to believe that these crystallins have evolved from the recruitment to the lens of preexisting metabolic enzymes, like ASL, by a process called 'gene sharing'.
[8] Mutations in the human ASL gene causes argininosuccinic aciduria, a rare autosomal recessive disorder, and results in deficiencies of the urea cycle.
Argininosuccinate lyase is an intermediate enzyme in the urea synthesis pathway and its function is imperative to the continuation of the cycle.
There is biochemical evidence that shows rises in ammonia can inhibit glutaminase and therefore limit the rate of synthesis of neurotransmitters such as glutamate,[11] which can explain the developmental delay in argininosuccinic aciduria patients.