Bcr-Abl tyrosine-kinase inhibitor
This abnormality was discovered by Peter Nowell in 1960[1] and is a consequence of fusion between the Abelson (Abl) tyrosine kinase gene at chromosome 9 and the break point cluster (Bcr) gene at chromosome 22, resulting in a chimeric oncogene (Bcr-Abl) and a constitutively active Bcr-Abl tyrosine kinase that has been implicated in the pathogenesis of CML.
While initial results have shown modest efficacy, further studies involving highly potent representatives of this drug class are necessary.
[6][7] Due to increasing resistance and intolerance to imatinib efforts were made to develop new drugs that could inhibit the Bcr-Abl tyrosine kinase.
[8] Imatinib (Gleevec) was discovered in 1992[9] and is regarded as first generation drug since it is the first Bcr-Abl tyrosine kinase inhibitor to be used in the treatment of CML.
[12] The shift of the AspPheGly (DFG) triad at the N-terminal end of the activation loop results in the exposure of a binding pocket which can be utilized by inhibitors.
Shortly after the introduction of imatinib, investigators began to describe a number of in vitro derived cell lines with resistance to the drug.
[4] Before a conclusion is drawn, it is important to consider that retrospective data has showed a high incidence of imatinib non-compliance in CML patients and this could lead to undesired clinical outcomes.
This substitution eliminates a critical oxygen molecule needed for hydrogen bonding between imatinib and the Abl kinase, and also creates steric hindrance to the binding of most TKIs.
[2] Ponatinib (Iclusig) by Ariad was approved in 2013 for use as second-line CML treatment, and is the only licensed TKI which binds to the T315I mutated kinase successfully.
[4] Some investigations in cell lines have shown that imatinib resistance may be partly due to an increase in the expression of the P-glycoprotein efflux pump.
Escalation of imatinib-doses has shown to overcome some cases of primary resistance to imatinib, such as Bcr-Abl duplication, but the response is usually short acting.
[2] There is also a growing interest in testing the hypothesis that administration of multiple Abl kinase inhibitors in early phase patients could be used to delay or prevent the emergence of drug resistant clones.
The combination of two agents targeting different pathways involved in CML may significantly improve response rates and potentially increase survival.
[13] Nilotinib binds to the inactive conformation of the Abl kinase domain, largely through lipophilic interactions and thus blocks its catalytic activity.
[18] Although nilotinib is more potent than imatinib it is possible that its specific mode of binding to Abl may make other sites vulnerable to new kinds of drug resistance.
[20] Although dasatinib is much more potent than imatinib it is possible, like with nilotinib, that its specific mode of binding to Abl may lead to new vulnerable sites that could confer new kinds of drug resistance.
[12][25] In contrast to imatinib, nilotinib and dasatinib, bosutinib is not an efficient substrate for multidrug resistance (MDR) transporters that promotes efflux of foreign molecules from cells.
[25] ARIAD Pharmaceuticals, Inc. announced on September 10, 2010 that ponatinib, an orally active Bcr-Abl TKI effective against the T315I mutation had been approved for a phase II clinical trial.
[27] Ariad used the highly potent drug lead, AP23464 to further investigate inhibitory possibilities of purine cored templates for dual Src/Abl inhibitors.
Then with in-vitro testing on inhibitory activity and in-vivo oral absorption assays a more lipophilic, amide bound, cyclopropyl group on C6 on the purine core was found to display both satisfactory pharmacokinetics and efficacy.
Finally modifications on the diarylamide side chain by adding imidazole appendages were inspired by then newly released nilotinib structure.
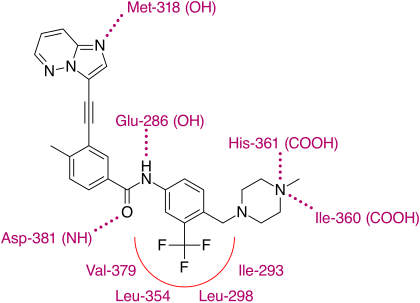
Imatinib has a terminal methyl piperazine group which has been shown to form a hydrogen bond with the carbonyl oxygen atom of residue Ile-360 in the activation loop of the Abl kinase.
Those speculations were confirmed with a two-fold increase in inhibitory action against Bcr-Abl T315I mutated kinase and the silver lining was the plasma protein binding of the substance (named '19a') appeared to have decreased, allowing for smaller doses with the same potency.
So, in attempts to reduce the molecule's lipophilicity further, substitution of a single carbon atom on the imidazo[1,2-a]pyridine core was made; which resulted in what is now known as the compound ponatinib.
[29] X-ray crystallographic analysis of ponatinib and T315I Bcr-Abl mutated kinase display that the imidazo[1,2b]pyridazine core rests in the adenine pocket of the enzyme.
[30] With this structure ponatinib has been shown to have a relatively broad kinase specificity profile which can probably be linked to the linearity of the linkage section of the molecule.
Attempts to utilize this pocket to increase efficacy led to the addition of various hydrophobic groups including single fluoro, bromo and chloro substituents.
Closer examination of the crystal structure of imatinib-kinase complex revealed Tyr-236 was in close proximity to the pyridine ring of imatinib, suggesting there was little or no room for a larger group there.
With that in mind a more hydrophilic pyrimidine ring was substituted for the pyridine, which was found to increase solubility while leaving efficacy the same or even slightly greater.
One Italian research group discovered through digital screening that commercially available thiadiazole derivatives displayed moderate inhibitory action on both Abl and Src kinases.