Docking (molecular)
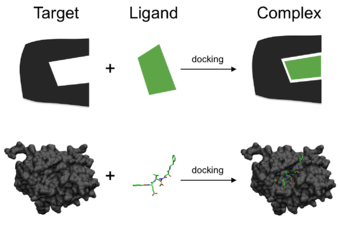
In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex.
[1] Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
The associations between biologically relevant molecules such as proteins, peptides, nucleic acids, carbohydrates, and lipids play a central role in signal transduction.
Molecular docking is one of the most frequently used methods in structure-based drug design, due to its ability to predict the binding-conformation of small molecule ligands to the appropriate target binding site.
Characterisation of the binding behaviour plays an important role in rational design of drugs as well as to elucidate fundamental biochemical processes.
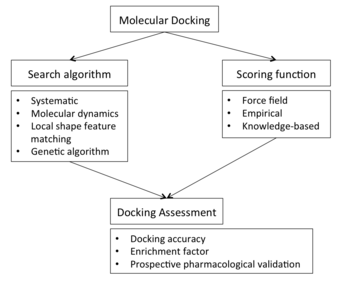
Molecular docking may be defined as an optimization problem, which would describe the “best-fit” orientation of a ligand that binds to a particular protein of interest.
The complementarity between the two surfaces amounts to the shape matching description that may help finding the complementary pose of docking the target and the ligand molecules.
[11][12][13] Whereas the shape complementarity based approaches are typically fast and robust, they cannot usually model the movements or dynamic changes in the ligand/protein conformations accurately, although recent developments allow these methods to investigate ligand flexibility.
Shape complementarity methods can quickly scan through several thousand ligands in a matter of seconds and actually figure out whether they can bind at the protein's active site, and are usually scalable to even protein-protein interactions.
Grid-based techniques, optimization methods, and increased computer speed have made docking simulation more realistic.
However, in practice with current computational resources, it is impossible to exhaustively explore the search space — this would involve enumerating all possible distortions of each molecule (molecules are dynamic and exist in an ensemble of conformational states) and all possible rotational and translational orientations of the ligand relative to the protein at a given level of granularity.
[24][25] Docking programs generate a large number of potential ligand poses, of which some can be immediately rejected due to clashes with the protein.
Most scoring functions are physics-based molecular mechanics force fields that estimate the energy of the pose within the binding site.
This gives a large number of false positive hits, i.e., ligands predicted to bind to the protein that actually don't when placed together in a test tube.
One way to reduce the number of false positives is to recalculate the energy of the top scoring poses using (potentially) more accurate but computationally more intensive techniques such as Generalized Born or Poisson-Boltzmann methods.
[9] The interdependence between sampling and scoring function affects the docking capability in predicting plausible poses or binding affinities for novel compounds.
[32] Docking screens can also be evaluated by the enrichment of annotated ligands of known binders from among a large database of presumed non-binding, “decoy” molecules.
[33] In the case of G protein-coupled receptors (GPCRs), which are targets of more than 30% of marketed drugs, molecular docking led to the discovery of more than 500 GPCR ligands.
For small molecules, several benchmark data sets for docking and virtual screening exist e.g. Astex Diverse Set consisting of high quality protein−ligand X-ray crystal structures,[35] the Directory of Useful Decoys (DUD) for evaluation of virtual screening performance,[29] or the LEADS-FRAG data set for fragments[36] An evaluation of docking programs for their potential to reproduce peptide binding modes can be assessed by Lessons for Efficiency Assessment of Docking and Scoring (LEADS-PEP).