Computational chemistry
[3] The complexity inherent in the many-body problem exacerbates the challenge of providing detailed descriptions of quantum mechanical systems.
[12] With the development of efficient computer technology in the 1940s, the solutions of elaborate wave equations for complex atomic systems began to be a realizable objective.
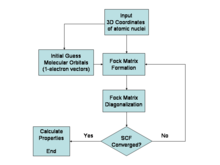
[15] The first ab initio Hartree–Fock method calculations on diatomic molecules were performed in 1956 at MIT, using a basis set of Slater orbitals.
The first configuration interaction calculations were performed in Cambridge on the EDSAC computer in the 1950s using Gaussian orbitals by Boys and coworkers.
[22] In 1964, Hückel method calculations (using a simple linear combination of atomic orbitals (LCAO) method to determine electron energies of molecular orbitals of π electrons in conjugated hydrocarbon systems) of molecules, ranging in complexity from butadiene and benzene to ovalene, were generated on computers at Berkeley and Oxford.
[29] Martin Karplus, Michael Levitt and Arieh Warshel received the 2013 Nobel Prize in Chemistry for "the development of multiscale models for complex chemical systems".
[37] Data that is difficult to obtain experimentally can be found using computational methods to model the mechanisms of catalytic cycles.
[38] Skilled computational chemists provide predictions that are close to experimental data with proper considerations of methods and basis sets.
With good computational data, researchers can predict how catalysts can be improved to lower the cost and increase the efficiency of these reactions.
[39] Methods like density functional theory can be used to model drug molecules and find their properties, like their HOMO and LUMO energies and molecular orbitals.
These computational simulations help researchers optimize the material find the best way to structure these nanomaterials before making them.
Purely calculated data avoids dealing with these adjusting for different experimental conditions like zero-point energy.
By continually improving these methods, scientists can get increasingly closer to perfectly predicting the behavior of atomic and molecular systems under the framework of quantum mechanics, as defined by the Schrödinger equation.
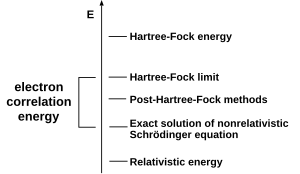
[51] To obtain exact agreement with the experiment, it is necessary to include specific terms, some of which are far more important for heavy atoms than lighter ones.
[52] A particularly important objective, called computational thermochemistry, is to calculate thermochemical quantities such as the enthalpy of formation to chemical accuracy.
Split operator methods help computers calculate these systems quickly by solving the sub problems in a quantum differential equation.
The database of compounds used for parameterization, i.e. the resulting set of parameters and functions is called the force field, is crucial to the success of molecular mechanics calculations.
The result of a molecular dynamics simulation is a trajectory that describes how the position and velocity of particles varies with time.
[68] Monte Carlo (MC) generates configurations of a system by making random changes to the positions of its particles, together with their orientations and conformations where appropriate.
Importance sampling methods are able to generate low energy states, as this enables properties to be calculated accurately.
This section focuses on the scaling of computational complexity with molecule size and details the algorithms commonly used in both domains.
In quantum chemistry, hybrid methods combining different computational approaches (like QM/MM) are increasingly used to tackle large biomolecular systems.
This intense computational demand arises from the inclusion of single and double excitations in the electron correlation calculation.
This elevated complexity restricts practical usage to smaller systems, typically up to 20-25 atoms in conventional implementations.
[91] An adaptation of the standard CCSD(T) method using local natural orbitals (NOs) to significantly reduce the computational burden and enable application to larger systems.
This advancement allows for practical applications to molecules of up to 100 atoms with reasonable basis sets, marking a significant step forward in computational chemistry's capability to handle larger systems with high accuracy.
In principle, it is possible to solve the Schrödinger equation in either its time-dependent or time-independent form, as appropriate for the problem in hand; in practice, this is not possible except for very small systems.
Significant errors can present themselves in ab initio models comprising many electrons, due to the computational cost of full relativistic-inclusive methods.
Present algorithms in computational chemistry can routinely calculate the properties of small molecules that contain up to about 40 electrons with errors for energies less than a few kJ/mol.
The treatment of larger molecules that contain a few dozen atoms is computationally tractable by more approximate methods such as density functional theory (DFT).