Exome sequencing
Exome sequencing is especially effective in the study of rare Mendelian diseases, because it is an efficient way to identify the genetic variants in all of an individual's genes.
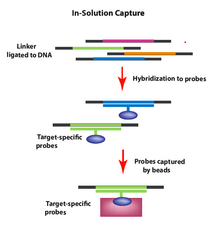
[citation needed] Target-enrichment methods allow one to selectively capture genomic regions of interest from a DNA sample prior to sequencing.
The preferred method is dependent on several factors including: number of base pairs in the region of interest, demands for reads on target, equipment in house, etc.
These 'short read' NGS systems are particularly well suited to analyse many relatively short stretches of DNA sequence, as found in human exons.
[citation needed] Exome sequencing is only able to identify those variants found in the coding region of genes which affect protein function.
A few strategies have been developed to improve the quality of exome data such as: Rare recessive disorders may not have single nucleotide polymorphisms (SNPs) in public databases such as dbSNP.
Incorporating these annotations can effectively boost the power of genetic association of rare variants analysis of whole genome sequencing studies.
With approaches such as exome sequencing, it is possible to significantly enhance the data generated from individual genomes which has put forth a series of questions on how to deal with the vast amount of information.
This additional depth makes exome sequencing well suited to several applications that need reliable variant calls.
[1][22] This heterogeneity of underlying risk means that very large sample sizes are required for gene discovery, and thus whole genome sequencing is not particularly cost-effective.
This sample size issue is alleviated by the development of novel advanced analytic methods, which effectively map disease genes despite the genetic mutations are rare at variant level.
[1] In Mendelian disorders of large effect, findings thus far suggest one or a very small number of variants within coding genes underlie the entire condition.
Because of the severity of these disorders, the few causal variants are presumed to be extremely rare or novel in the population, and would be missed by any standard genotyping assay.
A study published in September 2009 discussed a proof of concept experiment to determine if it was possible to identify causal genetic variants using exome sequencing.
They sequenced four individuals with Freeman–Sheldon syndrome (FSS) (OMIM 193700), a rare autosomal dominant disorder known to be caused by a mutation in the gene MYH3.
[2] This was the first reported study that used exome sequencing as an approach to identify an unknown causal gene for a rare mendelian disorder.
Subsequently, another group reported successful clinical diagnosis of a suspected Bartter syndrome patient of Turkish origin.
Exome sequencing revealed an unexpected well-conserved recessive mutation in a gene called SLC26A3 which is associated with congenital chloride diarrhea (CLD).
This example provided proof of concept of the use of whole-exome sequencing as a clinical tool in evaluation of patients with undiagnosed genetic illnesses.
They looked at variants that have the potential to be pathogenic such as non-synonymous mutations, splice acceptor and donor sites and short coding insertions or deletions.
Previous exome sequencing studies of common single nucleotide polymorphisms (SNPs) in public SNP databases were used to further exclude candidate genes.
This exciting finding demonstrates that exome sequencing has the potential to locate causative genes in complex diseases, which previously has not been possible due to limitations in traditional methods.
Targeted capture and massively parallel sequencing represents a cost-effective, reproducible and robust strategy with high sensitivity and specificity to detect variants causing protein-coding changes in individual human genomes.
Knowledge of this gene's function guided the infant's treatment, leading to a bone marrow transplantation which cured the child of disease.
[24] Researchers have used exome sequencing to identify the underlying mutation for a patient with Bartter Syndrome and congenital chloride diarrhea.
[12] Bilgular's group also used exome sequencing and identified the underlying mutation for a patient with severe brain malformations, stating "[These findings] highlight the use of whole exome sequencing to identify disease loci in settings in which traditional methods have proved challenging... Our results demonstrate that this technology will be particularly valuable for gene discovery in those conditions in which mapping has been confounded by locus heterogeneity and uncertainty about the boundaries of diagnostic classification, pointing to a bright future for its broad application to medicine".