Kallmann syndrome
[2][3][4] If left untreated, people will have poorly defined secondary sexual characteristics, show signs of hypogonadism, almost invariably are infertile and are at increased risk of developing osteoporosis.
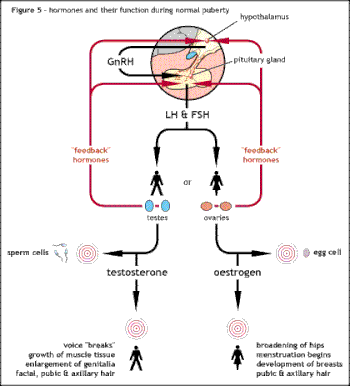
This leads to olfactory problems such as anosmia, optic defects like color blindness, and results in hypothalmic deficiencies associated with low levels of LH, affecting sex hormone testosterone in males or estrogen and progesterone in females.
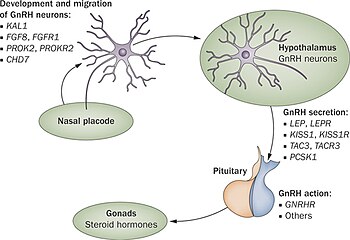
[12][13] Kallmann syndrome is caused by mutations in several genes involved in the development of the hypothalamus and olfactory bulbs, including KAL1, FGFR1, FGF8, PROKR2, and PROK2.
[14] It is normally difficult to distinguish a case of Kallmann syndrome (KS)/hypogonadotropic hypogonadism (HH) from a straightforward constitutional delay of puberty.
However, if puberty has not started by either age 14 (girls) or 15 (boys) years and one or more of the non-reproductive features mentioned below is present, then a referral to reproductive endocrinologist might be advisable.
The HPG axis then either fails totally or is reduced to a very low level of GnRH release in adult life with no obvious cause (e.g. a pituitary tumour).
[17][20] Functional hypothalamic amenorrhoea is seen in females where the HPG axis is suppressed in response to physical or psychological stress or malnutrition but is reversible with the removal of the stressor.
[8][4] Affected individuals with KS and other forms of HH are almost invariably born with normal sexual differentiation; i.e., they are physically male or female.
This is due to the human chorionic gonadotrophin (hCG) produced by placenta at approximately 12 to 20 weeks gestation (pregnancy) which is normally unaffected by having KS or CHH.
[21] People with KS/HH lack the surge of GnRH, LH, and FSH that normally occurs between birth and six months of age, referred to as mini-puberty.
It involves taking a specialised X-ray picture of the spine and hips and measuring the bone mineral density and comparing the result to the average value for a young healthy adult in the general population.
[25] To date at least 25 different genes have been implicated in causing Kallmann syndrome or other forms of hypogonadotropic hypogonadism through a disruption in the production or activity of GnRH (37).
It causes the x-linked form of Kallmann syndrome and is associated with the additional symptoms of anosmia, bimanual synkinesis and renal agenesis.
[8][3] The underlying cause of Kallmann syndrome or other forms of hypogonadotropic hypogonadism is a failure in the correct action of the hypothalamic hormone GnRH.
[29] The term hypogonadism describes a low level of circulating sex hormones; testosterone in males and oestrogen and progesterone in females.
[citation needed] In the first 10 weeks of normal embryonic development, the GnRH releasing neurons migrate from their original source in the nasal region and end up inside the hypothalamus.
[30] Diagnosing KS and other forms of CHH is complicated by the difficulties in distinguishing between a normal constitutional delay of puberty or a case of KS/CHH.
[38] Diagnosis of KS/CHH normal involves a range of clinical, biochemical and radiological tests to exclude other conditions that can cause similar symptoms.
[citation needed] [4][3] [4][3] [4][3] For both males and females, the initial aim for treatment is the development of the secondary sexual characteristics normally seen at puberty.
[3][39][34][35][40] Once this has been achieved, continued hormone replacement therapy is required for both males and females to maintain sexual function, bone health, libido and general wellbeing.
[3] In males, the monitoring of treatment normally requires the measurement of serum testosterone, inhibin B, haematocrit and prostate-specific antigen (PSA).
Ovulation induction can be achieved either with pulsatile GnRH therapy or alternatively with gonadotropin injections (hCG, FSH, hMG) given at set intervals to trigger the maturation and release of the egg for natural conception.
[44][45] The link between anosmia and hypogonadism was noted already in 1856 by the Spanish physician Aureliano Maestre de San Juan[12] who described a 40-year old male who, upon autopsy, exhibited absent olfactory bulbs, undeveloped testicles, micropenis, and lack of pubic hair.
[11] A 1961 case report by the Austrian pathologist Richard Ladislaus Heschl[11] noted an association between male hypogonadism (including an unmasculinised larynx,[46] and sparse body and pubic hair[11]) and anatomical absence of the olfactory nerves, bulb, and tract.
It was the case of a man aged 45, in all respects well developed, with the exception of the testicles, which were not larger than beans and contained no seminal canals, and the larynx, which seemed to be of feminine dimensions.
In the mucous membrane of the nose there was also an absence of nerves.In 1914, Franz Weidenreich performed autopsies on cadavers of 10 people who had had anosmia, uncovering hypogonadism in three and postulating a syndromic association.