Machine learning in bioinformatics
[2][3] Prior to the emergence of machine learning, bioinformatics algorithms had to be programmed by hand; for problems such as protein structure prediction, this proved difficult.
These methods contrast with other computational biology approaches which, while exploiting existing datasets, do not allow the data to be interpreted and analyzed in unanticipated ways.
Artificial neural networks in bioinformatics have been used for:[5] The way that features, often vectors in a many-dimensional space, are extracted from the domain data is an important component of learning systems.
), techniques such as principal component analysis are used to project the data to a lower dimensional space, thus selecting a smaller set of features from the sequences.
[14] Convolutional networks were inspired by biological processes[15][16][17][18] in that the connectivity pattern between neurons resembles the organization of the animal visual cortex.
This reduced reliance on prior knowledge of the analyst and on human intervention in manual feature extraction makes CNNs a desirable model.
[25] Computationally, random forests are appealing because they naturally handle both regression and (multiclass) classification, are relatively fast to train and to predict, depend only on one or two tuning parameters, have a built-in estimate of the generalization error, can be used directly for high-dimensional problems, and can easily be implemented in parallel.
Statistically, random forests are appealing for additional features, such as measures of variable importance, differential class weighting, missing value imputation, visualization, outlier detection, and unsupervised learning.
Clustering is central to much data-driven bioinformatics research and serves as a powerful computational method whereby means of hierarchical, centroid-based, distribution-based, density-based, and self-organizing maps classification, has long been studied and used in classical machine learning settings.
An example of a hierarchical clustering algorithm is BIRCH, which is particularly good on bioinformatics for its nearly linear time complexity given generally large datasets.
[35] Natural language processing and text mining have helped to understand phenomena including protein-protein interaction, gene-disease relation as well as predicting biomolecule structures and functions.
This trend began in 1951 when Pauling and Corey released their work on predicting the hydrogen bond configurations of a protein from a polypeptide chain.
[4][43] The current state-of-the-art in secondary structure prediction uses a system called DeepCNF (deep convolutional neural fields) which relies on the machine learning model of artificial neural networks to achieve an accuracy of approximately 84% when tasked to classify the amino acids of a protein sequence into one of three structural classes (helix, sheet, or coil).
[44] Currently, limitations and challenges predominate in the implementation of machine learning tools due to the amount of data in environmental samples.
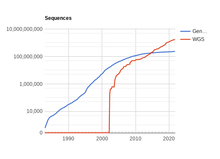
[46] Many algorithms were developed to classify microbial communities according to the health condition of the host, regardless of the type of sequence data, e.g. 16S rRNA or whole-genome sequencing (WGS), using methods such as least absolute shrinkage and selection operator classifier, random forest, supervised classification model, and gradient boosted tree model.
[48] In addition, random forest (RF) methods and implemented importance measures help in the identification of microbiome species that can be used to distinguish diseased and non-diseased samples.
The framework combines: They demonstrated performance by analyzing two published datasets from large-scale case-control studies: The proposed approach improved the accuracy from 81% to 99.01% for CDI and from 75.14% to 90.17% for CRC.
The most commonly used methods are radial basis function networks, deep learning, Bayesian classification, decision trees, and random forest.
[2] In addition, machine learning has been applied to systems biology problems such as identifying transcription factor binding sites using Markov chain optimization.
As proposed by Titano[58] 3D-CNN techniques were tested in supervised classification to screen head CT images for acute neurologic events.
Text Nailing, an alternative approach to machine learning, capable of extracting features from clinical narrative notes was introduced in 2017.
This technique has been applied to the search for novel drug targets, as this task requires the examination of information stored in biological databases and journals.
[74] Given their direct relationship to catalytic enzymes, and compounds produced from their encoded pathways, BGCs/GCFs can serve as a proxy to explore the chemical space of microbial secondary metabolism.
Cataloging GCFs in sequenced microbial genomes yields an overview of the existing chemical diversity and offers insights into future priorities.
In 2017, researchers at the National Institute of Immunology of New Delhi, India, developed RiPPMiner[76] software, a bioinformatics resource for decoding RiPP chemical structures by genome mining.
[80] antiSMASH allows the rapid genome-wide identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genomes.
[83] MIBiG facilitates the standardized deposition and retrieval of biosynthetic gene cluster data as well as the development of comprehensive comparative analysis tools.
It empowers next-generation research on the biosynthesis, chemistry and ecology of broad classes of societally relevant bioactive secondary metabolites, guided by robust experimental evidence and rich metadata components.
[84] SILVA[85] is an interdisciplinary project among biologists and computers scientists assembling a complete database of RNA ribosomal (rRNA) sequences of genes, both small (16S, 18S, SSU) and large (23S, 28S, LSU) subunits, which belong to the bacteria, archaea and eukarya domains.
[86] Greengenes[87] is a full-length 16S rRNA gene database that provides chimera screening, standard alignment and a curated taxonomy based on de novo tree inference.