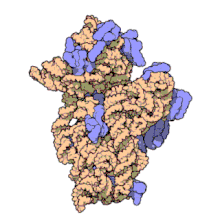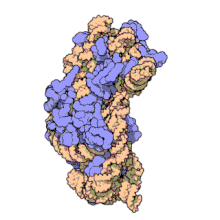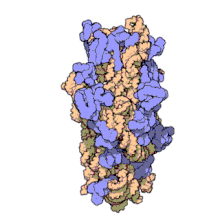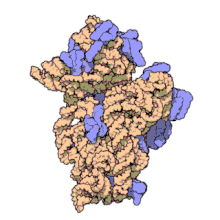
16S ribosomal RNA
[9] Some thermophilic archaea (e.g. order Thermoproteales) contain 16S rRNA gene introns that are located in highly conserved regions and can impact the annealing of "universal" primers.
[21][22] As a result, 16S rRNA gene sequencing has become prevalent in medical microbiology as a rapid and cheap alternative to phenotypic methods of bacterial identification.
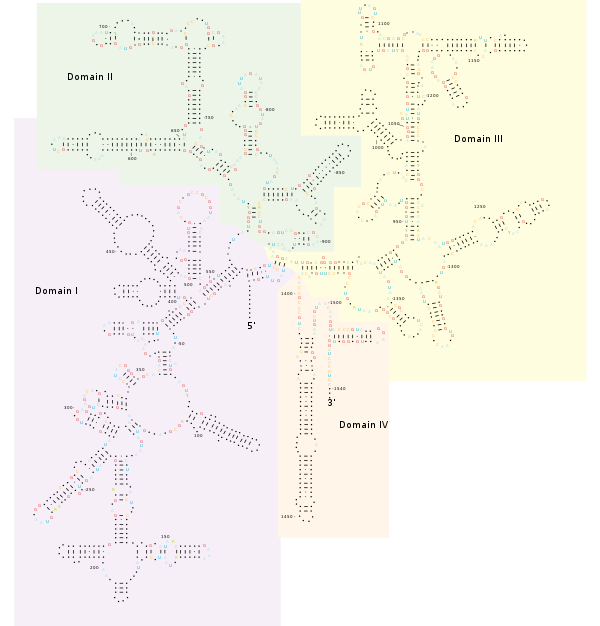
[29] The bacterial 16S gene contains nine hypervariable regions (V1–V9), ranging from about 30 to 100 base pairs long, that are involved in the secondary structure of the small ribosomal subunit.
[31] While the entire 16S sequence allows for comparison of all hypervariable regions, at approximately 1,500 base pairs long it can be prohibitively expensive for studies seeking to identify or characterize diverse bacterial communities.
[33] While 16S hypervariable regions can vary dramatically between bacteria, the 16S gene as a whole maintains greater length homogeneity than its eukaryotic counterpart (18S ribosomal RNA), which can make alignments easier.
[31] Many community studies select semi-conserved hypervariable regions like the V4 for this reason, as it can provide resolution at the phylum level as accurately as the full 16S gene.
[37] As a result, the V4 sequences can differ by only a few nucleotides, leaving reference databases unable to reliably classify these bacteria at lower taxonomic levels.
[37] By limiting 16S analysis to select hypervariable regions, these studies can fail to observe differences in closely related taxa and group them into single taxonomic units, therefore underestimating the total diversity of the sample.
[37] Under the assumption that evolution is driven by vertical transmission, 16S rRNA genes have long been believed to be species-specific, and infallible as genetic markers inferring phylogenetic relationships among prokaryotes.
In addition to observations of natural occurrence, transferability of these genes is supported experimentally using a specialized Escherichia coli genetic system.
[43] GreenGenes is a quality controlled, comprehensive 16S rRNA gene reference database and taxonomy based on a de novo phylogeny that provides standard operational taxonomic unit sets.
[46] EzBioCloud database, formerly known as EzTaxon, consists of a complete hierarchical taxonomic system containing 62,988 bacteria and archaea species/phylotypes which includes 15,290 valid published names as of September 2018.
It contains no redundancy, so only one representative for each species was considered avoiding same sequences from different strains, isolates or pathovars resulting in a very fast tool for microorganisms identification, compatible with any classification software (QIIME, Mothur, DADA, etc).