Ohtahara syndrome
The syndrome is outwardly characterized by tonic spasms and partial seizures within the first few months of life,[3] and receives its more elaborate name from the pattern of burst activity on an electroencephalogram (EEG).
It is an extremely debilitating progressive neurological disorder, involving intractable seizures and severe intellectual disabilities.
[4] Ohtahara syndrome is rare and the earliest-appearing age-related epileptic encephalopathy, with seizure onset occurring within the first three months of life, and often in the first ten days.
Many children die from OS within their first 2 years of life, while those who survive maintain physical and cognitive disabilities such as excessive fatigue, difficulty feeding, chest infections and slow developmental progress.
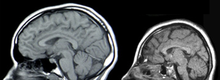
[24][2] Typically, onset of seizures and spasms have been indicative of OS diagnosis, while MRI and abnormal EEG "burst suppression" pattern can confirm.
Genetic testing with chromosomal microarray analysis followed by an epilepsy gene panel or whole exome sequencing may be considered after MRI imaging has been exhausted.
[citation needed] Prognosis is poor for infants with OS, and can be characterized by management of seizures, effects of secondary symptoms and shortened life span (up to 3 years of age).
Family members of infants with OS may consult with a palliative care team as symptoms may worsen or develop.
[8] Prospects of recovering from OS after hemispherectomy surgery has been shown to be favorable, with patients experiencing "catch up" in development.
Currently, only one clinical trial has been performed to examine the efficacy of high-definition (HD) transcranial direct-current stimulation (HD-tDCS) in reducing epileptiform activity.