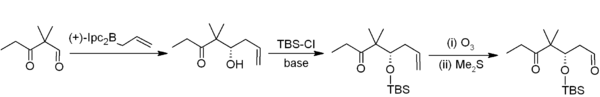Organoboron chemistry
[1][2] Organoboranes and -borates enable many chemical transformations in organic chemistry — most importantly, hydroboration and carboboration.
Most reactions transfer a nucleophilic boron substituent to an electrophilic center either inter- or intramolecularly.
Oxidation or protonolysis of the resulting organoboranes generates many organic products, including alcohols, carbonyl compounds, alkenes, and halides.
The trialkyl and triaryl derivatives feature a trigonal-planar boron center that is typically only weakly Lewis acidic.
When the alkyl group is small, such as methyl, the monoalkylboranes tend to redistribute to give mixtures of diborane and di- and trialkylboranes.
Trimethyl borate, debatably not an organoboron compound, is an intermediate in sodium borohydride production.
[19] Triethylborane adducts can be synthesised directly from the imidazolium salt and lithium triethylborohydride.
One example is the diborene (RHB=BHR):[20][21] Each boron atom has an attached proton and is coordinated to a NHC carbene.
Boronic acids RB(OH)2 react with potassium bifluoride K[HF2] to form trifluoroborate salts K[RBF3],[25] precursors to nucleophilic alkyl and aryl boron difluorides, ArBF2:[26] In hydroboration, alkenes insert into borane B-H bonds, with anti-Markovnikov stereochemistry.
Hydroboration with borane (BH3) equivalents converts only 33% of the starting olefin to product — boron-containing byproducts consume the remainder.
[27][28] Metal-catalyzed borylation reactions produce an organoboron compound from aliphatic or aromatic C-H sigma bonds via a transition-metal catalyst.
Carbon monoxide reacts with alkylboranes to form an unstable borane carbonyl.
For example, homologated primary alcohols result from organoboranes, carbon monoxide, and a reducing agent (here, sodium borohydride):[29] Alkynylboranes attack electrophiles to give trans alkenylboranes,[30] as in the first step of this olefin synthesis: The key property of organoboranes (R3B) and borates (R4B−, generated via addition of R− to R3B) is their susceptibility to reorganization.
The boron-attached carbon is nucleophilic;[31] in borates, the nucleophicity suffices for intermolecular transfer to an electrophile.
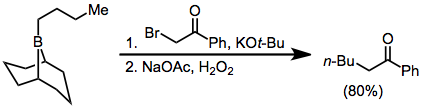
Consequently, organoboranes are easily removed from an alkane or alkene substrate, as in the second step of this olefin synthesis:[30] α-Halo enolates are common nucleophiles in borane reorganization.
After nucleophilic attack at boron, the resulting ketoboronate eliminates the halogen and tautomerizes to a neutral enolborane.
A functionalized carbonyl compound then results from protonolysis,[36] or quenching with other electrophiles: Because the migration is stereospecific, this method synthesizes enantiopure α-alkyl or -aryl ketones.
[37] α-Haloester enolates add similarly to boranes, but with lower yields:[38] Diazoesters and diazoketones remove the requirement for external base.
[40] In allylboration, an allylborane adds across an aldehyde or ketone with an allylic shift, and can then be converted to a homoallylic alcohol during workup.
[41] For example, in Nicolaou's epothilones synthesis, asymmetric allylboration (with an allylborane derived from chiral alpha-pinene) is the first step in a two-carbon homologation to acetogenin:[42] Trifluoroborate salts are stabler than boronic acids and selectively alkylate aldehydes:[43] The hydroboration-oxidation reaction pair oxidizes the borane to an alcohol with hydrogen peroxide or to a carbonyl group with chromium oxide.
With excess base, two of the three alkyl groups attached to the boron atom may convert to halide, but disiamylborane permits only halogenation of the hydroborated olefin:[45]Treatment of an alkenylborane with iodine or bromine induces migration of a boron-attached organic group.
Borane hydrides such as 9-BBN and L-selectride (lithium tri(sec-butyl)borohydride) are reducing agents.
Triethylborane was used to ignite the JP-7 fuel of the Pratt & Whitney J58 variable cycle engines powering the Lockheed SR-71 Blackbird.
Organoboron compounds have long been discussed for use as boron delivery agents in neutron capture therapy of cancer.