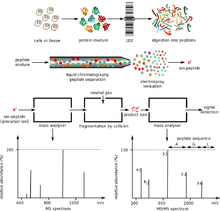
Protein mass spectrometry
(MALDI) Matrix-assisted laser desorption ionization was coined in the late 1980s by Franz Hillenkamp and Michael Karas.
In 1987, Koichi Tanaka used the "ultra fine metal plus liquid matrix method" and ionized biomolecules the size of 34,472 Da protein carboxypeptidase-A.
Around the same time MALDI became popularized, John Bennett Fenn was cited for the development of electrospray ionization.
[5][6] Koichi Tanaka received the 2002 Nobel Prize in Chemistry alongside John Fenn, and Kurt Wüthrich "for the development of methods for identification and structure analyses of biological macromolecules.
In electrospray, the ions are created from proteins in solution, and it allows fragile molecules to be ionized intact, sometimes preserving non-covalent interactions.
In MALDI, the proteins are embedded within a matrix normally in a solid form, and ions are created by pulses of laser light.
[8] Whole-protein mass analysis is primarily conducted using either time-of-flight (TOF) MS, or Fourier transform ion cyclotron resonance (FT-ICR).
Tandem mass spectrometry (MS/MS) is used to measure fragmentation spectra and identify proteins at high speed and accuracy.
Tandem MS of whole protein ions has been investigated recently using electron capture dissociation and has demonstrated extensive sequence information in principle but is not in common practice.
This approach is referred to as "top-down" strategy of protein analysis as it involves starting with the whole mass and then pulling it apart.
The top-down approach however is mostly limited to low-throughput single-protein studies due to issues involved in handling whole proteins, their heterogeneity and the complexity of their analyses.
[8] In the second approach, referred to as the "bottom-up" MS, proteins are enzymatically digested into smaller peptides using a protease such as trypsin.
Hence, this approach uses identification at the peptide level to infer the existence of proteins pieced back together with de novo repeat detection.
[9] The smaller and more uniform fragments are easier to analyze than intact proteins and can be also determined with high accuracy, this "bottom-up" approach is therefore the preferred method of studies in proteomics.
This is exacerbated by the fact that enzymatic digestion of a protein gives rise to a large number of peptide products.
In light of these problems, the methods of one- and two-dimensional gel electrophoresis and high performance liquid chromatography are widely used for separation of proteins.
In this dimension, the protein is separated by its isoelectric point (pI) and the second-dimension is SDS-polyacrylamide gel electrophoresis (SDS-PAGE).
If this information does not allow unequivocal identification of the protein, its peptides can be subject to tandem mass spectrometry for de novo sequencing.
De novo sequencing has proven successful for confirming and expanding upon results from database searches.
To this end, antigen presenting cells expose protein fragments via MHC molecules to the immune system.
However, the individual signal depends on the primary structure of the protein, on the complexity of the sample, and on the settings of the instrument.
[30] By using chemical crosslinking to couple parts of the protein that are close in space, but far apart in sequence, information about the overall structure can be inferred.
Parallel analysis of the genome and the proteome facilitates discovery of post-translational modifications and proteolytic events,[34] especially when comparing multiple species.