Saethre–Chotzen syndrome
Saethre–Chotzen syndrome (SCS), also known as acrocephalosyndactyly type III, is a rare congenital disorder associated with craniosynostosis (premature closure of one or more of the sutures between the bones of the skull).
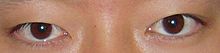
Individuals with SCS also have droopy eyelids (ptosis), widely spaced eyes (hypertelorism), and minor abnormalities of the hands and feet (syndactyly).
The majority of individuals with SCS are moderately affected, with uneven facial features and a relatively flat face due to underdeveloped eye sockets, cheekbones, and lower jaw.
In addition to the physical abnormalities, people with SCS also experience growth delays, which results in a relatively short stature.
More severe cases of SCS, with more serious facial deformities, occurs when multiple cranial sutures close prematurely.
However, the temporal and parietal bones are separated by sutures, which remain open, allowing the head to slightly change in shape during childbirth.
The TWIST gene encodes a basic helix-loop-helix (b-HLH) transcription factor that controls head mesenchyme development as the cranial tube forms.
Mice that were lacking both copies of the TWIST gene were spontaneously aborted prior to birth, and had serious deformities including abnormal limb and head defects and failure of the neural tube to properly close.
Since none of these mutations were seen in normal individuals who didn't have SCS, this provided enough evidence to conclude that the TWIST gene was the causative agent of SCS1.
[2] Prenatal diagnosis of Saethre-Chotzen Syndrome in high risk pregnancies is doable, but very uncommon and rarely performed.
Prenatal testing can also be performed during weeks 10–12 using chorionic villus sampling (CVS) to extract DNA from the fetus.
[7] Recently, there has been an increased interest in utilizing ultrasound equipment in order to detect fetal skull abnormalities due to immature fusion of the cranial sutures.
Sequence analysis of exon 1 (TWIST1 coding region) provides a good method for detecting the frequency of mutations in the TWIST1 gene.
Common methods include PCR, multiplex ligation-dependent probe amplification (MLPA), and chromosomal microarray (CMA).
Direct gene testing uses blood, hair, skin, amniotic fluid, or other tissues in order to find genetic disorders.
One of the common symptoms of SCS is the development of short (brachydactyly), webbed fingers and broad toes (syndactyly).
These characteristics do not cause any problems to the function of the hands or feet, and thus, no medical procedure is required to fix the abnormalities, unless the patient requests it.
[7] This is especially the case in individuals with asymmetrical unilateral coronal synostosis, which requires reconstructive surgery of the face and skull.
Vision problems usually arise due to a lack of space in the eye orbit and skull because of the abnormal bone structure of the face.
[2] Midfacial surgery may also be required during early childhood to correct respiratory problems, dental malocclusion, and swallowing difficulties.
A year later in 1932, F. Chotzen, a German psychiatrist, described a father and his two sons as having very similar characteristics as the mother and her daughters, as well as having hearing loss, short stature, and mild mental retardation.