Selective estrogen receptor modulator
[3] Compared to pure ER agonists–antagonists (e.g., full agonists and silent antagonists), SERMs are more tissue-specific, allowing them to selectively inhibit or stimulate estrogen-like action in various tissues.
Adverse effects of tamoxifen include hot flashes and an increase in the risk of developing endometrial cancer compared to women of similar age.
It shows no breast or endometrial stimulation and in the first two years the small increase is better in venous thromboembolism, and similar in the long term to other SERMs.
The advantage of bazedoxifene over raloxifene is that it increases endothelial nitric oxide synthase activity and does not antagonize the effect of 17β-estradiol on vasomotor symptoms.
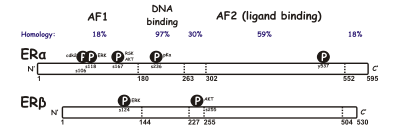
ERα is considered the main medium where estrogen signals are transduced at the transcriptional level and is the predominant ER in the female reproductive tract and mammary glands while ERβ is primarily in vascular endothelial cells, bone, and male prostate tissue.
[14] The ligand-binding domain is a globular, three-layered structure made of 11 helixes and contains a pocket for the natural or synthetic ligand.
[14][13] Influencing factors for binding affinity are mainly the presence of a phenol moiety, molecular size and shape, double bonds and hydrophobicity.
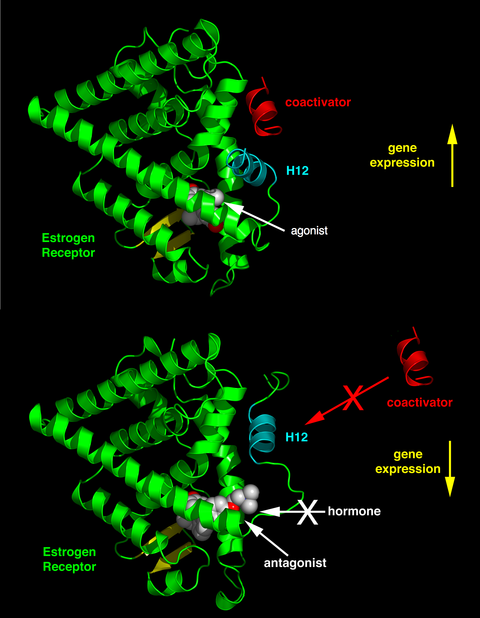
Helices 3, 5, and 12 together form a binding surface for an NR box motif contained in coactivators with the canonical sequence LXXLL (where L represents leucine or isoleucine and X is any amino acid).
There is growing evidence to support that SERM activity is mainly determined by selective recruitment of corepressors and coactivators to ER target genes in specific types of tissues and cells.
The ligand-binding domain of the ER demonstrates how ligands promote and prevent coactivator binding based on the shape of the estrogen or antiestrogen complex.
Post-translation modification of coactivators can result in a dynamic model of steroid hormone action by way of multiple kinase pathways initiated by cell surface growth factor receptors.
The ER substrates or coenzyme A can be polyubiquitinated by multiple cycles of the reaction or, depending on linkage proteins, they can either be activated further or degraded by the 26S proteasome.
This is likely due to the ethyl group of the tamoxifen stilbene core that is subject to allylic oxidative activation causing DNA alkylation and strand scission.
[9] Tamoxifen is more promiscuous than raloxifene in target sites because of the relationship between ER's amino acid in Asp-351 and the antiestrogenic side chain of the SERM.
[24] The structure and activity relationship of toremifene is similar to that of tamoxifen, but it has a substantial improvement from the older drug in regards to DNA alkylation.
The presence of the added chlorine atom reduces the stability of cations formed from activated allylic metabolites and thus decreases alkylation potential, and indeed toremifene does not display DNA adduct formation in rodent hepatocytes.
[26][31] Third-generation compounds display either no uterine stimulation, improved potency, no significant increases in hot flushes or a combination of these attributes.
[9] The first dihydronapthalene SERM, nafoxidine, was a clinical candidate for the treatment of breast cancer but had side effects including severe phototoxicity.
The amine-bearing side chain can then adopt an axial conformation and locate this group orthogonally to the plane of the core, like ralofoxifene and other less uterotropic SERMs.
[9] Unlike raloxifene, lasofoxifene satisfies the requirement of a pharmacophore model that predicts resistance to gut wall glucuronidation.
The structural requirement is a non-planar topology with the steric bulk close to the plane of a fused bicyclic aromatic system.
Lasofoxifene's large flexible side chain terminates in a pyrrolidine head group and threads its way out toward the surface of the protein, where it interferes directly with the positioning of the AF-2 helix.
Structure–activity relationship studies showed that by removing that group of tamoxifen agonistic activity in the uterus was significantly reduced, but not in bone and cardiovascular system.
[15] Other distinctive bindings to the ligand-binding pocket are with a nearly planar "core" structure typically composed of a biaryl heterocycle, equivalent to the A-ring and B-ring of 17β-estradiol, to the corresponding binding site; a bulky side chain from the biaryl structure, analogous to the B-ring of 17β-estradiol and finally a second side group that is the C- and D-ring equivalent and usually aromatic, fills the remainder volume of the ligand-binding pocket.
Amino acids in the ligand-binding domains differ at two positions, Leu-384 and Met-421 in ERα and Met-336 and Ile-373 in ERβ, but they have similar hydrophobicity and occupying volumes.
However, the shapes and the rotational barrier of the amino acid residues are not the same, leading to distinguish α- and β-face of the binding cavity between ERα and ERβ.
Many ERβ-selective ligands have a largely planar arrangement as the binding cavity of ERβ is slightly narrower than that of ERα, however, this by itself leads to modest selectivity.
[8] It was another ten years before tamoxifen was approved in December 1977, not as a contraceptive but as a hormonal treatment to treat and prevent breast cancer.
[9] Ospemifene was approved on February 26, 2013, for the treatment of moderate to severe dyspareunia, which is a symptom, due to menopause, of vulvar and vaginal atrophy.
Combined therapy with conjugated estrogens and the SERM bazedoxifene, was approved on October 3, 2013, for the treatment of vasomotor symptoms linked with menopause.