[4][5] Senapathy's theory cannot explain the strong evidence for common descent (homology, universal genetic code, embryology, fossil record.
)[6] Genes of all organisms, except bacteria, consist of short protein-coding regions (exons) interrupted by long sequences (introns).
It soon became apparent that a typical eukaryotic gene was interrupted at many locations by introns, dividing the coding sequence into many short exons.
introns in the gene It was also discovered that the spliceosome machinery was large and complex with ~300 proteins and several SnRNA molecules.
Regarding this requirement, Doolittle said, “It is extraordinarily difficult to imagine how informationally irrelevant sequences could be introduced into pre-existing structural genes without deleterious effects.” He stated “I would like to argue that the eukaryotic genome, at least in that aspect of its structure manifested as ‘genes in pieces’ is in fact the primitive original form.” James Darnell expressed similar views in 1978.
They speculated that the precellular stage had primitive RNA genes which had introns, which were reverse transcribed into DNA and formed the progenote.
In their view, introns originated as spacer sequences that enabled convenient recombination and shuffling of exons that encoded distinct functional domains in order to evolve new genes.
[19][20] Furthermore, molecular biologists questioned the exon shuffling proposal, from a purely evolutionary view for both methodological and conceptual reasons, and, in the long run, this theory did not survive.
In contrast, the coding sequence of eukaryotes existed only in short segments of exons of ~120 bases regardless of the length of the protein.
As random DNA sequences could be generated in the computer, Senapathy thought that he could ask these questions and conduct his experiments in silico.
Furthermore, when he began studying this question, sufficient DNA and protein sequence information existed in the National Biomedical Research Foundation (NBRF) database in the early 1980s.
The split genes thus originated from random DNA sequences by choosing the best of the short coding segments (exons) and splicing them.
These findings indicated that split genes could have originated from random DNA sequences with exons and introns as they appear in today's eukaryotic organisms.
The presence of a random sequence was therefore sufficient to create in the primordial ancestor the segregated form of RNA observed in the eukaryotic gene structure.
Thus, in response to evolutionary pressures to create larger and more complex genes, the RNA fragments were joined together by a splicing mechanism that removed the introns.
If this hypothesis was true, the split genes of today's living organisms should contain stop codons exactly at the ends of introns.
He took the data for 1,030 splice junction sequences (donors and acceptors) and counted the codons occurring at each of the 7- base positions in the donor signal sequence [CAG:GTGAGT] and each of the possible 2-base positions in the acceptor signal [CAG:G] from the GenBank database.
Nirenberg again stated that these observations fully supported the split gene theory for the origin of splice junction sequences from stop codons.
[2][24] Soon after the discovery of introns by Philip Sharp and Richard Roberts, it became known that mutations within splice junctions could lead to diseases.
[25] The final step of the splicing process occurs when the two exons are joined and the intron is released as a lariat RNA.
[26] Several investigators found the branch point sequences in different organisms[25] including yeast, human, fruit fly, rat, and plants.
Senapathy discovered that stop codons occur as key parts in every genetic element in eukaryotic genes.
Research based on the split gene theory sheds light on other basic questions of exons and introns.
The spliceosomal machinery would be required to remove them and to enable the short exons to be linearly spliced together as a contiguously coding mRNA that can be translated into a complete protein.
Thus, the split gene theory argues that spliceosomal machinery exists to remove the unnecessary introns.
It also suggests why a splicing mechanism was developed at the start of primordial evolution.”[16] Senapathy proposed a plausible mechanistic and functional rationale why the eukaryotic nucleus originated, a major question in biology.
These investigations thus led to the possibility that primordial DNA with essentially random sequence gave rise to the complex structure of the split genes with exons, introns and splice junctions.
Cells that harbored split genes had to be complex with a nuclear cytoplasmic boundary, and must have a spliceosomal machinery.
[1][2][3] According to the split gene theory, this process of intron loss could have happened from prebiotic random DNA.
Senapathy developed algorithms to detect donor and acceptor splice sites, exons and a complete split gene in a genomic sequence.
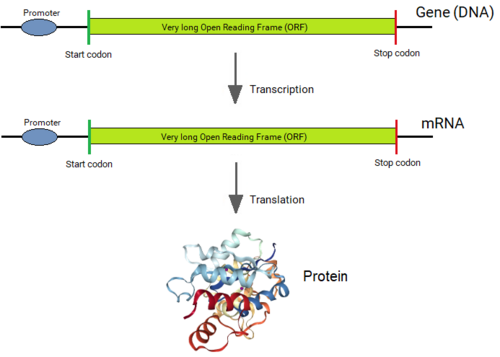
Transcription, splicing and translation of a eukaryotic gene.
A eukaryotic gene consists of a promoter, exons, introns and a poly-A addition site. It is transcribed into a primary RNA transcript (or pre-mRNA) by the RNA polymerase enzyme. This RNA undergoes the process of editing by the spliceosome for the precise removal of the introns and joining of the exons, which produces the mRNA molecule. This mRNA contains the complete coding sequence without any interrupting stop codons that is translated by the ribosome into the protein encoded by the gene. In the figure, the lengths of introns are short, but in reality, they are extremely long, on average 20 times longer than the exons, and often even much longer up to around 500,000 bases. The exons are usually short with an average of ~120 bases, and with an upper maximum of ~600 bases.
[
1
]
Also shown is an example protein structure (
PDB ID: 2VUX
) for the Human ribonucleotide reductase, subunit M2 B.
The clustering of stop codons in a random DNA sequence lead to rare ORFs that are long.
The negative exponential frequency distribution of the ORF lengths in a random sequence indicates that, in a linear sequence, the shorter the ORFs they are more frequent, and the longer the ORFs they are less and less frequent. Thus, there is a tendency for stop codons to be clustered in most places in a sequence, and, therefore, the longer ORFs are rarer, even within the upper maximum length of ~600 bases. Senapathy reasoned that, coding sequence segments from the available long ORFs could be chosen as exons, whereas the intervening sequences with clusters of stop codons could be earmarked as introns to be deleted from the primary RNA transcript, which would lead to a split gene structure.
The negative exponential distribution of ORF lengths in a random DNA sequence and in eukaryotic DNA sequences.
Senapathy found that stop codons occur at a high frequency in a random DNA sequence, as 3 stop codons exist out of 64 codons, which leads to short open reading frames (ORFs) with average length of ~60 bases. He also found that the lengths of the ORFs are distributed in a negative exponential manner. This plot indicates that the frequency of the zero ORF length (consecutive stop-codons occurring tandemly) is the most frequent of all ORF lengths, the frequency of ORF length of one codon (3 bases) is the next most frequent, and so on. The frequency of longer ORFs reduces exponentially, and reaches a zero frequency around an ORF length of ~600 bases, which means that ORFs longer than 600 bases do not occur.
[
1
]
Surprisingly, the plot from the eukaryotic DNA sequences was almost exactly the same as that from the random DNA sequences.
Corroboration of the Split Gene Theory by actual DNA sequences of human genes.
The Split Gene Theory predicts that all three stop codons should be present at a high frequency in each of the three reading frames (RFs), which would lead to short Open Reading Frames (ORFs). It also predicts that: a) exons would occur within these short ORFs in all three RFs; b) introns would be long, and that c) exon lengths would be confined to ORF lengths. These predictions are precisely true in the DNA sequences of most eukaryotic genes. Two example genes (
FLJ35894
and
ADCY1
) from the human genome are shown. All of the exons in each gene are short, and most of the introns are long. In each gene, the exons (short yellow boxes) are confined to short ORFs that occurred in the DNA sequence. In addition, stop codons occur at the ends of the exons, which are actually part of the splice junction sequences.
Origin of splice junction sequences from the stop codons.
[
1
]
[
2
]
[
3
]
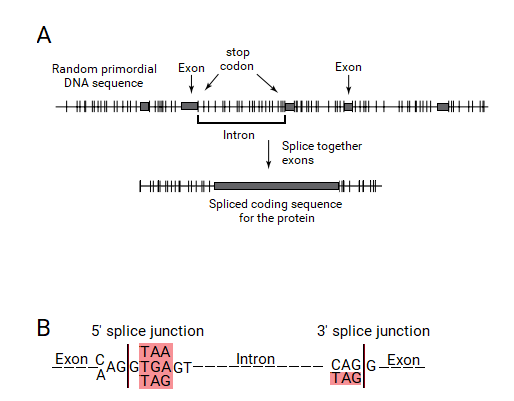
(A) The molecular machinery that chose the exons of a split gene from a random primordial DNA sequence should be capable of searching for stop codons (tick marks) to identify regions without stop codons (in the primary RNA copy, not shown), which are the ORFs. In doing so, the first encountered stop codon will be marked as the start of the intron. This process will lead to the presence of a stop codon at the beginning of the introns. Sometimes, all of an Open Reading Frame is chosen to be an exon, because of which the end of the previous intron will have a stop codon. (B) The start and the end of the intron are parts of the "splice junction sequences," that signal the exact point of splicing to the spliceosome machinery. The stop codons are shown with a red background.
Stop codons occur as key parts of all important genetic elements in eukaryotic genes.
The key genetic elements of eukaryotic genes are the promoters, donor and acceptor splice junction signals, lariat (branch point) signals, and poly-A addition sites. The core component of each of these genetic elements is a stop codon.
Origin of bacterial genes from split genes.
The split genes of modern eukaryotes with short exons (average length of 120 bases, and maximum of ~600 bases) interrupted by long introns are extremely probable in random DNA sequences due to the reasons described in the section
Origin of introns and the split gene structure
, above. In contrast, the long contiguously coding bacterial genes (that can be as long and 10,000 bases, and longer up to 90,000 bases) without introns are practically impossible to occur in random sequences. Thus, the only way that bacterial genes could originate was to delete the introns from the split genes that occurred in random DNA sequences and produce contiguously coding genes. The example protein shown with its 3D structure is from PDB database (
ID:1UNF
).