Comparative genomics
[5] In comparative genomics, synteny is the preserved order of genes on chromosomes of related species indicating their descent from a common ancestor.
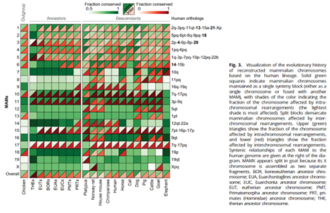
[12] Comparisons of genome synteny between and within species have provided an opportunity to study evolutionary processes that lead to the diversity of chromosome number and structure in many lineages across the tree of life;[13][14] early discoveries using such approaches include chromosomal conserved regions in nematodes and yeast,[15][16] evolutionary history and phenotypic traits of extremely conserved Hox gene clusters across animals and MADS-box gene family in plants,[17][18] and karyotype evolution in mammals and plants.
The system helps researchers to identify large rearrangements, single base mutations, reversals, tandem repeat expansions and other polymorphisms.
[29] At the same time, Bonnie Berger, Eric Lander, and their team published a paper on whole-genome comparison of human and mouse.
Instead of undertaking their own analyses, most biologists can access these large cross-species comparisons and avoid the impracticality caused by the size of the genomes.
These methods can also quickly uncover single-nucleotide polymorphisms, insertions and deletions by mapping unassembled reads against a well annotated reference genome, and thus provide a list of possible gene differences that may be the basis for any functional variation among strains.
Based on a variety of biological genome data and the study of vertical and horizontal evolution processes, one can understand vital parts of the gene structure and its regulatory function.
It is however often complicated by the multiplicity of events that have taken place throughout the history of individual lineages, leaving only distorted and superimposed traces in the genome of each living organism.
[32][33] Comparative genomics plays a crucial role in identifying copy number variations (CNVs) and understanding their significance in evolution.
CNVs, which involve deletions or duplications of large segments of DNA, are recognized as a major source of genetic diversity, influencing gene structure, dosage, and regulation.
While single nucleotide polymorphisms (SNPs) are more common, CNVs impact larger genomic regions and can have profound effects on phenotype and diversity.
Ongoing research aims to address these questions using techniques like comparative genomic hybridization, which allows for a detailed examination of CNVs and their significance.
[36] Comparative genomics holds profound significance across various fields, including medical research, basic biology, and biodiversity conservation.
[37][38][39] To tackle this challenge, comparative genomics offers a solution by pinpointing nucleotide positions that have remained unchanged over millions of years of evolution.
These conserved regions indicate potential sites where genetic alterations could have detrimental effects on an organism's fitness, thus guiding the search for disease-causing variants.
Moreover, comparative genomics holds promise in unraveling the mechanisms of gene evolution, environmental adaptations, gender-specific differences, and population variations across vertebrate lineages.
[41] For instance, in animal genetics, indigenous cattle exhibit superior disease resistance and environmental adaptability but lower productivity compared to exotic breeds.
[44] Computational approaches will remain critical for research and teaching, especially when information science and genome biology is taught in conjunction.
Additionally, ongoing efforts focus on optimizing existing algorithms to handle the vast amount of genome sequence data by enhancing their speed.
It integrates elements of colinear sequence alignment and gene orthology prediction, presenting a greater challenge due to the vast size and intricate nature of whole genomes.
Despite its complexity, numerous methods have emerged to tackle this problem because WGAs play a crucial role in various genome-wide analyses, such as phylogenetic inference, genome annotation, and function prediction.
Analysis based on coalescence theory tries predicting the amount of time between the introduction of a mutation and a particular allele or gene distribution in a population.
Identifying the loci of advantageous genes is a key step in breeding crops that are optimized for greater yield, cost-efficiency, quality, and disease resistance.
Previous methods of identifying loci associated with agronomic performance required several generations of carefully monitored breeding of parent strains, a time-consuming effort that is unnecessary for comparative genomic studies.
[78] In May 2019, using the Global Genome Set, a team in the UK and Australia sequenced thousands of globally-collected isolates of Group A Streptococcus, providing potential targets for developing a vaccine against the pathogen, also known as S.
Because of their morphological, physiological, and genetic resemblance to humans, mice and rats have long been the preferred species for biomedical research animal models.
In order to comprehend its TCRs and their genes, Glusman conducted research on the sequencing of the human and mouse T cell receptor loci.
Comparisons of the genomic sequences within each physical site or location of a specific gene on a chromosome (locs) and across species allow for research on other mechanisms and other regulatory signals.
Some suggest new hypotheses about the evolution of TCRs, to be tested (and improved) by comparison to the TCR gene complement of other vertebrate species.
A comparative genomic investigation of humans and mice will obviously allow for the discovery and annotation of many other genes, as well as identifying in other species for regulatory sequences.