Structural variation
Originally, a structure variation affects a sequence length about 1kb to 3Mb, which is larger than SNPs and smaller than chromosome abnormality (though the definitions have some overlap).
The first study in 2004 that used DNA microarrays could detect tens of genetic loci that exhibited copy number variation, deletions and duplications, greater than 100 kilobases in the human genome.
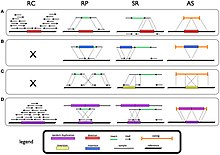
[3][4] These structural variants include deletions, tandem duplications, inversions, mobile element insertions.
In recent studies, copy-number variations are tested on people who do not have genetic diseases, using methods that are used for quantitative SNP genotyping.
Because CNVs are usually caused by unequal recombination, widespread similar sequences such as LINEs and SINEs may be a common mechanism of CNV creation.
[3] Other classes of complex structural variant include deletion-inversion-deletions, duplication-inversion-duplications, and tandem duplications with nested deletions.
A complete comparison between human and chimpanzee structural variation also suggested that some of these may be fixed in one species because of its adaptative function.
In that system, both "inner" and "outer" coordinates are shown; they are both not actual breakpoints, but surmised minimal and maximum range of sequence affected by the structural variation.
[citation needed] New methods have been developed to analyze human genetic structural variation at high resolutions.
The best established of the PCR based methods is real time quantitative polymerase chain reaction (qPCR).
[30] An SNP genotyping method that offers independent fluorescence intensities for two alleles can be used to target the nucleotides in between two copies of a segmental duplication.