Vitamin B12 total synthesis
The total synthesis of the complex biomolecule vitamin B12 was accomplished in two different approaches by the collaborating research groups of Robert Burns Woodward at Harvard[1][2][3][4][5] and Albert Eschenmoser at ETH[6][7][8][9][10][11][12] in 1972.
[35] The second model synthesis, published 1969,[36] explored a novel photochemical cycloisomerization process to create the direct A/D-ring junction as final corrin-ring closure between rings A and D.[37] The A/B approach to the cobyric acid syntheses was collaboratively pursued and accomplished in 1972 at Harvard.
[41][17]: 29 Woodward's recognition of the stereochemical enigma that came to light by the irritating behavior of one of his carefully planned synthetic steps became, according to his own writings,[41] part of the developments that led to the orbital symmetry rules.
[39][43] Since independent progress of the two groups towards their long-term objective was so clearly complementary, Woodward and Eschenmoser decided in 1965[18]: 1497 [17]: 30 to join forces and to pursue from then on the project of a B12 synthesis collaboratively, planning to utilize the ligand construction (ring coupling of components) strategy of the ETH model system.
This newly developed method turned out to provide a general solution to the problem of constructing the characteristic structural elements of the corrin chromophore, the vinylogous amidine systems bridging the four peripheral rings.
In both approaches, the four peripheral rings derived from enantiopure precursors possessing the correct sense of chiral, thereby circumventing major stereochemical problems in the buildup of the ligand system.
Eschenmoser had discussed the ETH contributions to the A/B approach in 1968 at the 22nd Robert A. Welch Foundation conference in Houston,[7] as well as in his 1969 RSC Centenary Lecture "Roads to Corrins", published in 1970.
[13]: 9,38 [note 11] Representative reviews of the two approaches to the chemical synthesis of vitamin B12 have been published in detail by A. H. Jackson and K. M. Smith,[45] T. Goto,[68] R. V. Stevens,[38] K. C. Nicolaou & E. G. Sorensen,[15][19] summarized by J. Mulzer & D. Riether,[69] and G. W. Craig,[14][33] besides many other publications where these epochal syntheses are discussed.
For this determination, the levo-rotatory ("unnatural") enantiomer of aminoketone (−)-H-3 was used in order to save precious material: Acylation of the amino group of (−)-H-3 with chloroacetyl chloride, followed by treatment of the product H-3a with potassium t-butoxide in t-butanol, afforded tetracyclic keto-lactame H-3b.
[1]: 527-528 [note 14] Coupling of ring-A and ring-D precursors to "pentacyclenone" N-acylation of tricyclic aminoketone (+)-H-3 with the chloride H-14 of carboxylic acid H-13 gave amide H-15, which on treatment with potassium t-butoxide in t-butanol stereoselectively produced pentacyclic keto-lactam H-16 via an intramolecular Michael reaction which directs the indicated hydrogen atoms in trans relationship to each other.
[1]: 539-540 [4]: 1:01:56-1:19:47 The construction of the corrin chromophore with its three vinylogous amidine units constitutes – besides the direct single bond connection between the rings A and D – the central challenge to any attempt to synthesize vitamin B12.
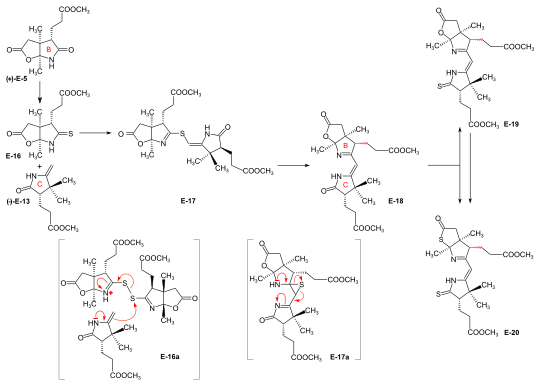
[2]: 287-290 [4]: 1:26:59-1:32:00 Induced by potassium t-butoxide in THF/t-butanol under rigorously controlled conditions with strict exclusion of air and moisture, the model A-D-component H-34u smoothly reacted with the B-C-component E-19[48]: 53-58 to give the sulfur-bridged coupling product HE-35u, named "thioether type I", in essentially quantitative yield.
[2]: 290 Starting with such mixtures of coupling products, at ETH a variety of conditions (e.g. methyl-mercury complex, BF3, triphenylphosphine[48]: 58-65 [2]: 291 ) were found to induce (via HE-38u) the contraction step to HE-39u in moderate yields.
[7]: 25-28 [8]: 387-389 [18]: 1563 In the explorations of ring-closure procedures for the much more highly substituted A/B-seco-corrinoid intermediate HE-39u, the ETH group focused on the intramolecular version of the oxidative sulfide contraction method, eventually leading to the dicyano-cobalt(III)-complex HE48u.
[2]: 304 The major obstacle in achieving an A/B-corrin-ring closure was the exposure of the highly unstable ring B exocyclic methylidene double bond, which tends to isomerize into a more stable, unreactive endocyclic position with great ease.
The product HE-45u was subjected to treatment with dimethylamine (as in the ETH variant), forming the highly labile methylidene derivative HE-46u, which then was converted with anhydrous CoCl2 in THF to dicyano-cobalt(III) complex HE-47u, the substrate ready to undergo the (A⇒B)-ring closure by a thioiminoester/enamine condensation.
[2]: 300 Since in corrin model syntheses such a C,C-condensation required induction by a strong base, its application in a substrate containing seven methylester groups was not without problems;[18]: 1562 in a, milder reactions conditions were applied.
Due to the high configurational lability of C-H chirogenic centers C-3, C-8 and C-13[4]: 1:21:49-1:23:42,1:35:43-1:36:14,1:51:51-1:52:30 at the ligand periphery in basic or acidic milieu, separation by HPLC was indispensable for isolation, purification and characterization of pure diastereomers of this and the following corrinoid intermediates.
[6]: 194-197 [8]: 380-386 [18]: 1537-1538 Conversion of lactam (+)-E-5 into the corresponding thiolactam E-16 (P2S5),[46]: 20-23,74-75 oxidation of E-16 with benzoyl peroxide in the presence of ring-C precursor (−)-E-13 (prepared at Harvard by the Cornforth route[note 6]), followed by heating the reaction product E-17 in triethylphosphite (as both solvent and thiophile) afforded B-C-component E-18 as a (not separated) mixture of two epimers (regarding the configuration of the propionic side chain at ring B) in up to 80 % yield.
This reaction concept developed at this stage, dubbed sulfide contraction,[6]: 199 [47][18]: 1534-1541 [37]: 1927-1941 turned out to make possible the construction of all three meso-carbon bridges of the vitamin's corrin ligand in both approaches of the synthesis.
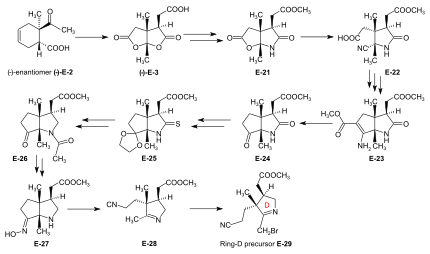
[61]: 114-116 Conventional conditions of an Arndt-Eistert reaction (SOCl2: acid chloride, then CH2N2 in THF: diazoketone, treated with Ag2O in MeOH) led to an – unforeseen, yet useful – ring closure of the originally formed chain-elongated ester through participation of the cyano group as a neighboring electrophile, affording the bicyclic enamino-ester derivative E-23.
[51]: 89-97 To attach ring-A precursor E-31, the ring B of E-32 was induced to expose its exocyclic methylidene double bond by treatment with dimethylamine in MeOH (using the method[note 19] developed by Schneider[48]: 32-34 ) forming E-33[51]: 108-115 which was subjected to the following cascade of operations:[51]: 130-150 iodination (N-iodosuccinimide, CH2Cl2, 0°), coupling with the thiolactam sulfur of the ring-A precursor E-31 [(CH3)3Si]2N-Na in benzene/t-BuOH), complexation (Cd(ClO4)2 in MeOH), treatment with triphenylphosphine/CF3COOH in boiling benzene (sulfide contraction) and, finally, re-complexation with Cd(ClO4)2/N,N-diisopropylethylamine in benzene/MeOH).
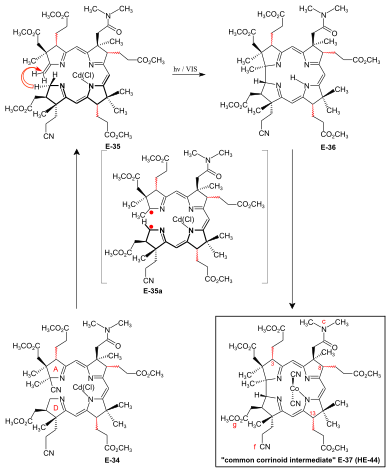
[51]: 39 [3]: 148-150 In the application of this novel process in the A/D approach of the cobyric acid synthesis,[9]: 86-95 [51]: 39-53 [12]: 1419 the reaction proceeded most efficiently and with highest coil stereoselectivity in favor of the natural A/D-trans junction in an A/D-seco-corrin cadmium complex.
[51]: 173 [12]: 1419 Corrin E-36 was immediately complexed (CoCl2,[18]: 1499-1500,1563-64 KCN, air, H2O, CH2Cl2) and finally isolated (thick-layer chromatography) as mixture of peripheral epimers in 45-50 % yield over four operations:[51]: 169-179 the common corrinoid intermediate dicyano-cobalt(III)-complex E-37 ≡ HE-44.
[51]: 42-43 Product mixtures from several such cycloisomerizations were combined for preparative HPLC separation and full characterization of the 14 isolated diastereomers of E-37[51]: 207-251 (of 16 theoretically possible, regarding helicity and the epimeric centers C-3, C-8, C-13[51]: 39 ).
These conceptually simple finishing steps turned out to be rather complex in execution, including unforeseen pitfalls like a dramatic loss of precious synthetic material in the so-called "Black Friday" (July 9, 1971).
[9]: 96-99 [55]: 19 [3]: 167 [18]: 1567-1568 In this final phase of the synthesis, HPLC again turned out to be absolutely indispensable for separation, isolation, characterization and, above all, identification of pure isomers of dicyano-cobalt(III)-complexes of totally as well as partially synthetic origin.
[55]: 19-21,39-43,146-205 [3]: 167-169 For steric reasons, only the predominant[55]: 19 [63]: 24 [4]: 2:08:20-2:09:02 C-3 α-epimer (with the C-3 side chain below the plane of the corrin ring) reacted to a 5,15-disubstituted product E-38/H-45, the reaction thus amounting to a chemical separation of the C-3 epimers.
[55]: 136-141 [3]: 170 In the remaining steps of the synthesis, only epimerization at C-13 played an important role,[55]: 19-21 with 13α being the configuration of the natural corrinoids, and 13β known as neo-epimers of vitamin B12 and its derivatives;[3]: 169-170 [81] these are readily separable by HPLC.
They also reciprocally provided structure proof for a specific constitutional isomer isolated from a mixture of isomeric mono-amides formed in the partial ammonolysis of the B12-derived cobester,[note 9] tentatively assigned to be the 3α,8α,13α-f-amide E-42/HE-49 (see fig.