Autosomal dominant polycystic kidney disease
[1] It is also the most common of the inherited cystic kidney diseases — a group of disorders with related but distinct pathogenesis, characterized by the development of renal cysts and various extrarenal manifestations, which in case of ADPKD include cysts in other organs, such as the liver, seminal vesicles, pancreas, and arachnoid membrane, as well as other abnormalities, such as intracranial aneurysms and dolichoectasias, aortic root dilatation and aneurysms, mitral valve prolapse, and abdominal wall hernias.
[1][5] ADPKD is estimated to affect at least one in every 1000 individuals worldwide, making this disease the most common inherited kidney disorder with a diagnosed prevalence of 1:2000 and incidence of 1:3000-1:8000 in a global scale.
[1] Although evidence exists for a two-hit mechanism (germline and somatic inactivation of two PKD alleles) explaining the focal development of renal and hepatic cysts,[15][16] haploinsufficiency is more likely to account for the vascular manifestations of the disease.
[17][18] Additionally, new mouse models homozygous for PKD1 hypomorphic alleles 22 and 23 and the demonstration of increased renal epithelial cell proliferation in PKD2 +/− mice suggest that mechanisms other than the two-hit hypothesis also contribute to the cystic phenotype.
[19] The significant intrafamilial variability observed in the severity of renal and extrarenal manifestations points to genetic and environmental modifying factors that may influence the outcome of ADPKD, and results of an analysis of the variability in renal function between monozygotic twins and siblings support the role of genetic modifiers in this disease.
[1][20] It is estimated that 43–78% of the variance in age to ESRD could be due to heritable modifying factors,[21][22] with parents as likely as children to show more severe disease in studies of parent-child pairs.
[34] However, molecular diagnostics can be necessary in the following situations: 1- when a definite diagnosis is required in young individuals, such as a potential living related donor in an affected family with equivocal imaging data;[34] 2- in patients with a negative family history of ADPKD, because of potential phenotypic overlap with several other kidney cystic diseases;[34] 3- in families affected by early-onset polycystic kidney disease, since in this cases hypomorphic alleles and/or oligogenic inheritance can be involved;[34][35] and 4- in patients requesting genetic counseling, especially in couples wishing a pre-implantation genetic diagnosis.
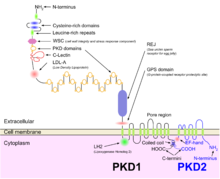
The large size and complexity of PKD1 and PKD2 genes, as well as marked allelic heterogeneity, present obstacles to molecular testing by direct DNA analysis.
[citation needed] Currently, the only pharmacological treatment available for ADPKD consists in reducing the speed in gain of total kidney volume (TKV) with vasopressin receptor 2 (V2) antagonists (i.e.
[citation needed] Recent research suggests that ketogenic dietary interventions beneficially affect the progression and symptoms in individuals with ADPKD.
[9][10] That study showed a significant decrease in the ratio of TKV increase and deterring of renal function decline in ADPKD patients after treatment with tolvaptan;[9][45] however, because laboratory test results regarding liver function appeared elevated in a percentage of patients enrolled in that study, the approval of the drug was either delayed by regulatory agencies or, as in case of the US, altogether denied.
[47] The mechanism was shown to involve the metabolic state of ketosis, and beneficial effects could be produced by time-restricted feeding, acute fasting, a ketogenic diet, or by supplementation with the ketone beta-hydroxybutyrate in mouse, rat and cat models of ADPKD.
[54] A retrospective case series study showed that ADPKD disease symptoms - including pain, hypertension and renal function - improved among 131 patients who implemented ketogenic diets for an average duration of 6 months.
[57][59] Dietary intake of oxalate or inorganic phosphate has been shown to accelerate PKD disease progression in several rodent models.
[57] Low levels or urinary citrate – a natural antagonist of the formation of harmful crystals in kidney tubules – have been shown to associate with worse disease progression in ADPKD patients.
[68] Many ADPKD patients experience symptomatic sequelae in consequence of the disease, such as cyst hemorrhage, flank pain, recurrent infections, nephrolithiasis, and symptoms of mass effect (i.e., early satiety, nausea and vomiting, and abdominal discomfort), from their enlarged kidneys.
[75] Epidemiological data shows that ADPKD affects 5–13.4% of patients undergoing hemodialysis in Europe and in the United States,[76][77][78] and about 3% in Japan.
[81][44] In this prognostic method, patients are divided into five subclasses of estimated kidney growth rates according to age-specific HtTKV ranges (1A, <1.5%; 1B, 1.5–3.0%; 1C, 3.0–4.5%; 1D, 4.5–6.0%; and 1E, >6.0%) as delineated in the CRISP study.