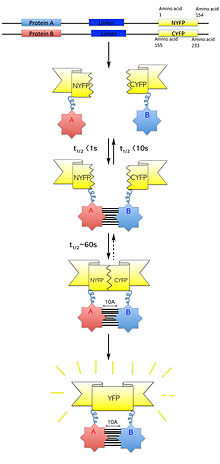
Bimolecular fluorescence complementation
[2] Therefore, through the visualisation and analysis of the intensity and distribution of fluorescence in these cells, one can identify both the location and interaction partners of proteins of interest.
Biochemical complementation was first reported in subtilisin-cleaved bovine pancreatic ribonuclease, then expanded using β-galactosidase mutants that allowed cells to grow on lactose.
[6] In 2000, Ghosh et al developed a system that allowed a green fluorescent protein (GFP) to be reassembled using an anti-parallel leucine zipper in E. coli cells.
The successful fluorescent signal indicated that the separate GFP peptide fragments were able to correctly reassemble and achieve tertiary folding.
It was, therefore, postulated that using this technique, fragmented GFP could be used to study interaction of protein–protein pairs that have their N–C termini in close proximity.
After the demonstration of successful fluorescent protein fragment reconstitution in mammalian cells, Hu et al. described the use of fragmented yellow fluorescent protein (YFP) in the investigation of bZIP and Rel family transcription factor interactions.
Transient gene expression is used to identify protein–protein interactions in vivo as well as in subcellular localisation of the BiFC complex.
[11] Reported linker sequences RSIAT and RPACKIPNDLKQKVMNH (single amino acid code) and AAANSSIDLISVPVDSR (Sigma) have been successfully used in BiFC experiments.
[14] Some controls include fluorophore fragments linked to non-interacting proteins, as the presence of these fusions tend to decrease non-specific complementation and false positive results.
During visualisation, one determines the fluorescence intensities of the BiFC complex and the internal control which, after subtracting background signal, becomes a ratio.
[11] Once the fusion proteins and controls have been designed and generated in their appropriate expression system, the plasmids must be transfected into the cells to be studied.
[15] The BiFC technology has been refined and expanded to include the abilities to simultaneously visualise multiple protein complexes in the same cell, RNA/protein interactions, to quickly detect changes in gene transduction pathways, demonstrate hidden phenotypes of drugs, where the predicted treatment outcome (i.e. cell death, differentiation, morphological change) is not seen in vivo, study complex formation in different cellular compartments, and to map protein interaction surfaces[13][19][20][21][22] The fluorescent signal only is produced after the proteins have interacted, which is generally in the order of hours.
The delay for chemical reactions to generate fluorophore may also have an effect on the dynamics of complex dissociation and partner exchange.
[1][7][8][23] BiFC complex formation is only reversible during the initial step of fluorescent reporter protein re-assembly, typically in the order of milliseconds.
Generally, this limitation is mitigated by ensuring that the fusion proteins of interest are expressed at endogenous concentrations.
Therefore, absence of fluorescence complementation may be a false negative and does not necessarily prove that the interaction in question does not occur.
[26] Certain fungi, such as Candida albicans, also have a high autofluorescent background, but BiFC can often still be performed when the proper controls and strains are used.
This problem can be alleviated by using structural information and the location of interaction sites to rationally identify fusion sites on the proteins of interest, using appropriate controls, and comparing the expression levels and functions of the fusion and wild-type proteins through Western Blots and functional assays.
Thus, the events of subunit joining signaled by the appearance of BiFC is an easy way to monitor ribosome biogenesis in contrast to laborious polysome profiling methods.
The fluorescent protein fragments used in BiFC have been expanded to include the colours blue, cyan, green, yellow, red, cherry, and Venus.
[13] BiFC has been expanded to include the study of RNA-binding protein interactions in a method Rackham and Brown described as trimolecular fluorescence complementation (TriFC).
Also known as the RNA bridge method, as the fluorophore and other interacting proteins form a bridge between the protein and the RNA of interest, this allows a simple detection and localisation of RNA-protein interactions within a living cell and provides a simple method to detect direct or indirect RNA-protein association (i.e. within a complex) that can be verified through in vitro analysis of purified compounds or RNAi knockdown of the bridging molecule(s).
[18][19] BiFC has been used to study nuclear translocation, via complex localisation, as well as interactions involving integral membrane proteins.
This allows protein–protein interaction surfaces to be mapped through the introduction of site-directed or random mutations that affect complex formation.
[39][40][41][42] Finally, by studying proteins in vitro, one is unable to determine the influence of specific protein–protein interactions in the cell on the functional or physiological consequences.
[43][44] Reversible fluorophore interaction Decreased sensitivity[45] Irreversible photo-bleaching[46][47][48] Erroneous transcription activation[14] Genetic complementation[4][5] Yeast as model organism[44][49] Overexpression of proteins[16] Nuclear localisation[16]