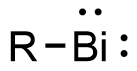Bismuthinidene
[1] Due to the unusually low valency and oxidation state of +1, most bismuthinidenes are reactive and unstable,[2] though in recent decades, both transition metals and polydentate chelating Lewis base ligands have been employed to stabilize the low-valent bismuth(I) center through steric protection and π donation either in solution or in crystal structures.
[6][7][8][9] These methods generally leveraged the ability of simple bismuth(I) halides or methylbismuth to ligate to tungsten, manganese, and chromium carbonyl complexes.
[6][7][10] One of the first examples of a monomeric bismuthinidene was discovered by Balasz et al., who used R = 2-(dimethylaminomethyl)phenyl to chelate a Bi(I) center through a combination of strong C-Bi and weak N-Bi interactions.
[11] Although the molecule quickly formed a cyclic oligomer, upon reaction with two equivalents of tungsten pentacarbonyl, monomeric crystalline RBi[W(CO)5]2 was isolated.
[3] This complex was first synthesized by reacting the precursor molecule LBiIIICl2 with two equivalents of the reducing agent K[B(iBu)3H] to yield isolable crystals of stable [C6H3-2,6-(C(Me)=N-2′,6′-Me2C6H3)2]Bi.
[12] In addition, calculated nucleus-independent chemical shift indices (NICS) and anisotropy of current-induced density (ACID) analysis show that the BiC3N ring of the molecule was stabilized by a certain degree of aromatic character due and may be classified as a benzazabismole to the delocalization of six π electrons, despite the nominally dative Bi-N bond.
[2] Phenylbismuth dichloride, stabilized by a diethyl/diisopropylphenyl-substituted cyclic alkyl amino carbene (Et2CAAC), reacts with one equivalent of the beryllium(0) complex Be(Et2CAAC)2 in toluene to give stable, isolable red crystals of the carbene-stabilized bismuthinidene (Et2CAAC)Bi-Ph.
In practice, both N,C,N- and N,C-chelated bismuthinidenes lose much of their Lewis acidic character due to nN → p*Bi donor-acceptor interactions.
However, the Lewis basicity of bismuthinidenes, particularly Dostál's N,C,N-coordinated bismuthinidene, allows them to cycle predictably between stable Bi(I) and Bi(III) oxidation states depending on the reaction conditions, allowing them to act as catalysts for a variety of different reactions, including transfer hydrogenations, deoxygenations, hydrodefluorinations, and dihydrogen reduction.
[14][15][21][25][26] In addition, bismuthinidenes react intrinsically with certain alkyl halides, dichalcogenides, and alkynes to form Bi(III) species.
[15][26] In 2019, Wang et al., who leveraged the catalytic activity of Dostál's bismuthinidene to catalyze a transfer hydrogenation reaction between ammonia-borane and azoarenes to form the corresponding arylhydrazines with good functional group tolerance.
[26] The reaction's catalytic cycle proceeds through the oxidative addition of two hydrogen atoms from ammonia-borane to the bismuth(I) center, forming a highly unstable bismuthine intermediate.
[26][31] Subsequent reductive elimination transfers the two hydrogen atoms across the pi bond of an azoarene molecule, restoring the bismuthinidene and forming arylhydrazine.
[14] When Dostál's bismuthinidene is exposed to gaseous N2O, the reaction mixture changes color from green to yellow and evolves dinitrogen gas.
The unstable Bi(III) hydride then undergoes aryl C-H reductive elimination, regenerating Phebox-Bi(I) and the hydrodefluorinated product.
[16] In 2019, Kořenková et al. discovered that Dostál's bismuthinidene behaves as a masked heterocyclic diene in the presence of the electron-deficient alkyne dimethyl acetylenedicarboxylate (DMAD), performing a hetero Diels-Alder [4+2] cycloaddition reaction to yield CO2Me-disubstituted 1-bisma-1,4-dihydro-iminonaphthalene, effectively converting one of the pendant imine arms of the bismuthinidene into a nitrogen-bridged bismacyclohexadiene, with the bismuth(III) atom serving as a bridgehead and the second imine arm largely losing coordination with the bismuth(III) center.
These complexes show significant covalent interaction between the bismuth(I) atoms and the cobalt or manganese centers, though these bismuth-metal bonds are dative in character.
Addition of this metastable bismuthinidene to THF solutions of M(CO)5 (where M = Cr, Mo, W) yields isolable crystals of [Ar’BiM(CO)5].