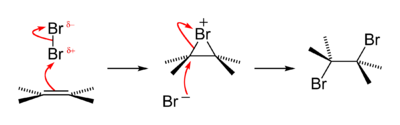Bromine compounds
The HBr/H2O system also involves many hydrates HBr·nH2O for n = 1, 2, 3, 4, and 6, which are essentially salts of bromine anions and hydronium cations.
Anhydrous hydrogen bromide is a poor solvent, only able to dissolve small molecular compounds such as nitrosyl chloride and phenol, or salts with very low lattice energies such as tetraalkylammonium halides.
The exceptions are decidedly in the minority and stem in each case from one of three causes: extreme inertness and reluctance to participate in chemical reactions (the noble gases, with the exception of xenon in the very unstable XeBr2; extreme nuclear instability hampering chemical investigation before decay and transmutation (many of the heaviest elements beyond bismuth); and having an electronegativity higher than bromine's (oxygen, nitrogen, fluorine, and chlorine), so that the resultant binary compounds are formally not bromides but rather oxides, nitrides, fluorides, or chlorides of bromine.
[4] Another method is halogen exchange in the presence of excess "halogenating reagent", for example:[4] When a lower bromide is wanted, either a higher halide may be reduced using hydrogen or a metal as a reducing agent, or thermal decomposition or disproportionation may be used, as follows:[4] Most of the bromides of the pre-transition metals (groups 1, 2, and 3, along with the lanthanides and actinides in the +2 and +3 oxidation states) are mostly ionic, while nonmetals tend to form covalent molecular bromides, as do metals in high oxidation states from +3 and above.
[4] The halogens form many binary, diamagnetic interhalogen compounds with stoichiometries XY, XY3, XY5, and XY7 (where X is heavier than Y), and bromine is no exception.
[5] Bromine monochloride (BrCl), a red-brown gas, quite readily dissociates reversibly into bromine and chlorine at room temperature and thus also cannot be obtained pure, though it can be made by the reversible direct reaction of its elements in the gas phase or in carbon tetrachloride.
[4] Bromine monofluoride in ethanol readily leads to the monobromination of the aromatic compounds PhX (para-bromination occurs for X = Me, But, OMe, Br; meta-bromination occurs for the deactivating X = –CO2Et, –CHO, –NO2); this is due to heterolytic fission of the Br–F bond, leading to rapid electrophilic bromination by Br+.
It reacts violently with water and explodes on contact with flammable materials, but is a less powerful fluorinating reagent than chlorine trifluoride.
It reacts vigorously with boron, carbon, silicon, arsenic, antimony, iodine, and sulfur to give fluorides, and will also convert most metals and many metal compounds to fluorides; as such, it is used to oxidise uranium to uranium hexafluoride in the nuclear power industry.
Bromine trifluoride is a useful nonaqueous ionising solvent, since it readily dissociates to form BrF+2 and BrF−4 and thus conducts electricity.
It also reacts violently with water and is a very strong fluorinating agent, although chlorine trifluoride is still stronger.
When bromine dissolves in aqueous solution, the following reactions occur:[10] Hypobromous acid is unstable to disproportionation.
[13] More important are the bromates, which are prepared on a small scale by oxidation of bromide by aqueous hypochlorite, and are strong oxidising agents.
Today, perbromates are produced by the oxidation of alkaline bromate solutions by fluorine gas.
The Br–O bond in BrO−4 is fairly weak, which corresponds to the general reluctance of the 4p elements arsenic, selenium, and bromine to attain their group oxidation state, as they come after the scandide contraction characterised by the poor shielding afforded by the radial-nodeless 3d orbitals.
Formally, compounds with this functional group may be considered organic derivatives of the bromide anion.