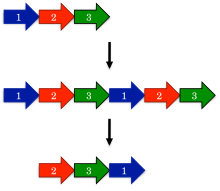
Circular permutation in proteins
Circular permutation can occur as the result of evolutionary events, posttranslational modifications, or artificially engineered mutations.
Fission and fusion occurs when partial proteins fuse to form a single polypeptide, such as in nicotinamide nucleotide transhydrogenases.
Circular permutations are routinely engineered in the laboratory to improve their catalytic activity or thermostability, or to investigate properties of the original protein.
[5] In 1989, Karolin Luger and her colleagues introduced a genetic method for making circular permutations by carefully fragmenting and ligating DNA.
Chris Ponting and Robert Russell identified a circularly permuted version of a saposin inserted into plant aspartic proteinase, which they nicknamed swaposin.
[7] Hundreds of examples of protein pairs related by a circular permutation were subsequently discovered in nature or produced in the laboratory.
[10] SISYPHUS is a database that contains a collection of hand-curated manual alignments of proteins with non-trivial relationships, several of which have circular permutations.
Saposins are highly conserved glycoproteins, approximately 80 amino acid residues long and forming a four alpha helical structure.
[17] It belongs to the saposin-like protein family (SAPLIP) and has the N- and C- termini "swapped", such that the order of helices is 3-4-1-2 compared with saposin, thus leading to the name "swaposin".
[20] These are membrane-bound enzymes that catalyze the transfer of a hydride ion between NAD(H) and NADP(H) in a reaction that is coupled to transmembrane proton translocation.
They consist of three major functional units (I, II, and III) that can be found in different arrangement in bacteria, protozoa, and higher eukaryotes.
[34] Two examples of frequently used methods that have problems correctly aligning proteins related by circular permutation are dynamic programming and many hidden Markov models.
[34] As an alternative to these, a number of algorithms are built on top of non-linear approaches and are able to detect topology-independent similarities, or employ modifications allowing them to circumvent the limitations of dynamic programming.
[36] Sequence methods are generally fast and suitable for searching whole genomes for circularly permuted pairs of proteins.
[37] They are often slower than sequence-based methods, but are able to detect circular permutations between distantly related proteins with low sequence similarity.
[38] This article was adapted from the following source under a CC BY 4.0 license (2012) (reviewer reports): Spencer Bliven; Andreas Prlić (2012).