Duchenne muscular dystrophy
[2] Initially, muscle loss occurs in the thighs and pelvis, extending to the arms,[3] which can lead to difficulties in standing up.
[3] Some individuals may experience intellectual disability,[3] and females carrying a single copy of the mutated gene may show mild symptoms.
[3] Duchenne muscular dystrophy is caused by mutations or deletions in any of the 79 exons encoding the large dystrophin protein, which is essential for maintaining the muscle fibers' cell membrane integrity.
[3] Diagnosis can frequently be made at birth through genetic testing, and elevated creatine kinase levels in the blood are indicative of the condition.
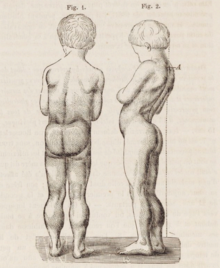
[15] They tend to walk on their toes,[15] in part due to shortening of the Achilles tendon,[16] and because it compensates for knee extensor weakness.
Another characteristic sign of Duchenne muscular dystrophy is pseudohypertrophy (enlarging) of the muscles of the tongue, calves, buttocks, and shoulders (around age 4 or 5).
Lumbar hyperlordosis is thought to be a compensatory mechanism in response to gluteal and quadriceps muscle weakness, all of which cause altered posture and gait (e.g.: restricted hip extension).
Mutations can either be inherited or occur spontaneously during germline transmission,[citation needed] causing a large reduction or absence of dystrophin, a protein that provides structural integrity in muscle cells.
[23] Dystrophin is responsible for connecting the actin cytoskeleton of each muscle fiber to the underlying basal lamina (extracellular matrix), through a protein complex containing many subunits.
The key tests performed on the biopsy sample for Duchenne muscular dystrophy are immunohistochemistry, immunocytochemistry, and immunoblotting for dystrophin, and should be interpreted by an experienced neuromuscular pathologist.
[31] Over the past several years, DNA tests have been developed that detect more of the many mutations that cause the condition, and muscle biopsy is not required as often to confirm the presence of Duchenne muscular dystrophy.
[33] Before invasive testing, determination of the fetal sex is important; while males are sometimes affected by this X-linked disease, female Duchenne muscular dystrophy is extremely rare.
[46][47] The antisense oligonucleotide golodirsen (Vyondys 53) was approved for medical use in the United States in 2019, for the treatment of cases that can benefit from skipping exon 53 of the dystrophin transcript.
[50] Casimersen (Amondys 45) was approved for medical use in the United States in February 2021,[52] and it is the first FDA-approved targeted treatment for people who have a confirmed mutation of the Duchenne muscular dystrophy gene that is amenable to exon 45 skipping.
[52] Comprehensive multidisciplinary care guidelines for Duchenne muscular dystrophy have been developed by the US Centers for Disease Control and Prevention and were published in 2010.
[53][54] Delandistrogene moxeparvovec (Elevidys) is a gene therapy that in June 2023 received United States FDA accelerated approval for the treatment of four and five-year-old children.
[55][56] In October 2023, the US Food and Drug Administration (FDA) approved Vamorolone (Agamree) as a Treatment for Duchenne muscular dystrophy.
[57] In March 2024, the US Food and Drug Administration (FDA) approved givinostat (Duvyzat), an oral medication, to be used in the treatment of Duchenne muscular dystrophy in people aged six years and older.
Givinostat is the first nonsteroidal drug to receive FDA approval for the treatment of all genetic variants of Duchenne muscular dystrophy.
In rare cases, people with Duchenne muscular dystrophy have been seen to survive into their forties or early fifties, with proper positioning in wheelchairs and beds, and the use of ventilator support (via tracheostomy or mouthpiece), airway clearance, and heart medications.
[63] Early planning of the required supports for later-life care has shown greater longevity for people with Duchenne muscular dystrophy.
[64] Curiously, in the mdx mouse model of Duchenne muscular dystrophy, the lack of dystrophin is associated with increased calcium levels and skeletal muscle myonecrosis.
[85] Antisense oligonucleotides (oligos), structural analogs of DNA, are the basis of a potential treatment for 10% of people with Duchenne muscular dystrophy.
[90] In 1990 England et al. noticed that a patient with mild Becker muscular dystrophy was lacking 46% of his coding region for dystrophin.
[91] Kole demonstrated success using splice-targeted AONs to correct missplicing in cells removed from beta-thalassemia patients[92][93] Wilton's group tested exon skipping for muscular dystrophy.
[94][95] Researchers are working on a gene editing method to correct a mutation that leads to Duchenne muscular dystrophy (DMD).
[102] Several medications designed to address the root cause are under development, including gene therapy and antisense drugs.
[2] Physical therapy, orthopedic braces, and corrective surgery may help with some symptoms[2] while assisted ventilation may be required in those with weakness of breathing muscles.