Hemoglobin E
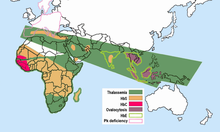
Hemoglobin E is very common among people of Southeast Asian, Northeast Indian, Sri Lankan and Bangladeshi descent.
[1][2] The βE mutation affects β-gene expression creating an alternate splicing site in the mRNA at codons 25-27 of the β-globin gene.
Also, this hemoglobin variant has a weak union between α- and β-globin, causing instability when there is a high amount of oxidant.
People who have hemoglobin E trait (heterozygous) are asymptomatic and their state does not usually result in health problems.
[7] Symptoms of hemoglobin E/β-thalassemia vary but can include growth retardation, enlargement of the spleen (splenomegaly) and liver (hepatomegaly), jaundice, bone abnormalities, and cardiovascular problems.
[8] Recommended course of treatment depends on the nature and severity of the symptoms and may involve close monitoring of hemoglobin levels, folic acid supplements, and potentially regular blood transfusions.
[4] This disease was first described by Virginia Minnich in 1954, who discovered a high prevalence of it in Thailand and initially referred to it as "Mediterranean Anaemia.
"[8] Hemoglobin E is most prevalent in mainland Southeast Asia (Thailand, Myanmar, Cambodia, Laos, Vietnam[9]), Sri Lanka, Northeast India and Bangladesh.