Hi-C (genomic analysis technique)
[1][2][3][4] Hi-C comprehensively detects genome-wide chromatin interactions in the cell nucleus by combining 3C and next-generation sequencing (NGS) approaches and has been considered as a qualitative leap in C-technology (chromosome conformation capture-based technologies) development and the beginning of 3D genomics.
[2][3][4] Similar to the classic 3C technique, Hi-C measures the frequency (as an average over a cell population) at which two DNA fragments physically associate in 3D space, linking chromosomal structure directly to the genomic sequence.
[4] The relative abundance of these chimeras, or ligation products, is correlated to the probability that the respective chromatin fragments interact in 3D space across the cell population.
[4] While 3C focuses on the analysis of a set of predetermined genomic loci to offer “one-versus-some” investigations of the conformation of the chromosome regions of interest, Hi-C enables “all-versus-all” interaction profiling by labeling all fragmented chromatin with a biotinylated nucleotide before ligation.
[3][4] As a result, biotin-marked ligation junctions can be purified more efficiently by streptavidin-coated magnetic beads, and chromatin interaction data can be obtained by direct sequencing of the Hi-C library.
[7] In recent years, Hi-C has found its application in a wide variety of biological fields, including cell growth and division, transcription regulation, fate determination, development, autoimmune disease, and genome evolution.
[4] At its inception, Hi-C was a low-resolution, high-noise technology that was only capable of describing chromatin interaction regions within a bin size of 1 million base pairs (Mb).
[9] Nevertheless, Hi-C data offered new insights for chromatin conformation as well as nuclear and genomic architectures, and these prospects motivated scientists to put efforts to modify the technique over the past decade.
[4] Standard Hi-C gives data on pairwise interactions at the resolution of 1 to 10 Mb, requires high sequencing depth and the protocol takes around 7 days to complete.
[4] Cells are lysed on ice with cold hypotonic buffer containing sodium chloride, Tris-HCl at pH 8.0, and non-ionic detergent IGEPAL CA-630, supplemented with protease inhibitors.
[4][16] This 5’ overhang provides the template required by the Klenow fragment of DNA Polymerase I to add biotinylated CTP or ATP to the digested ends of chromatin.
[4][16] Since this ligation step occurs between blunt-ended DNA fragments (since the sticky ends have been filled in with biotin-labeled bases), the reaction is allowed to go on for up to 4 hours to make up for its inherent inefficiency.
[4][16] This is achieved by using a combination of enzymes that fill in 5’ overhangs, and add 5’ phosphate groups and adenylate to the 3’ ends of fragments to allow for ligation of sequencing adaptors.
[20] These variants, in addition to others (described below), represent modifications to the foundational technique of standard Hi-C and address and alleviate one or more limitations of the original method.
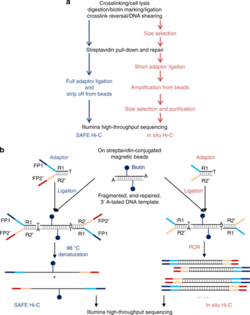
[17] SAFE Hi-C has been demonstrated to increase library complexity due to the removal of PCR duplicates which lower the overall percentage of unique paired reads.
[24][25] Hsieh et al. analyzed 2.64 billion reads from mouse embryonic stem cells and demonstrated that there was increased power for detecting short-range interactions.
[24][25][26]Hi-C has also been adapted for use with single cells but these techniques require high levels of expertise to perform and are plagued with issues such as low data quality, coverage, and resolution.
[28] The chimeric DNA ligation products generated by Hi-C represent pairwise chromatin interactions or physical 3D contacts within the nucleus,[1][2][3][4] and can be analyzed by a variety of downstream approaches.
[29][30] Reads mapped more than the maximum molecule length away from the closest restriction sites are the results of physical breakage of the chromatin or non-canonical nuclease activities.
[29] Because these reads also instruct information on chromatin interactions, they are not discarded, but appropriate filtering must take place after assigning genomic locations to remove technical noise in the dataset.
For example, potential undigested restriction sites could be specifically filtered out, rather than passively identified, by removing reads mapped to the same chromosomal strand with a small distance (user-defined, experience-based) in between.
Li et al. in 2018 described deDoc, a method where bin size is selected as the one at which the structural entropy of the Hi-C matrix reaches a stable minimum.
[29] Various polymer models[54][55] exist to statistically characterize the properties of loci pairs separated by a given distance, but discrete binning and fitting continuous functions are two common ways to analyze the distance-dependent interaction frequencies between datapoints.
[61] Variations of this approach with different objective functions, such as Lavaburst,[62] MrTADFinder,[63] 3DNetMod,[64] and Matryoshka,[65] are also developed to achieve better computing performance on higher resolution datasets.
[29][30] Instead, point mutations are identified as outliers with higher interaction frequencies than expected within the Hi-C matrix, given that the background model consists only of the strongest signals such as the distance-decay functions.
Due to the ability of Hi-C to depict dynamic interactions in differentiation-related TADs, the researchers discovered increases in the number of DHS sites, CTCF binding ability, active histone modifications, and target gene expressions within these TADs of interest, and found significant participation of major pluripotency factors such as OCT4, NANOG, and SOX2 in the interaction network during somatic cell reprogramming.
[11] Since then, Hi-C has been recognized as one of the standard methods to probe for transcriptional regulatory activities, and has confirmed that chromosome architecture is closely related to cell fate.
Kloetgen et al. used in situ Hi-C to study T cell acute lymphoblastic leukemia (T-ALL) and found a TAD fusion event that removed a CTCF insulation site, allowing for the oncogene MYC’s promoter to directly interact with a distal super enhancer.
[80] Fang et al. have also shown how there are T-ALL specific gain or loss of chromatin insulation, which alters the strength of TAD architecture of the genome, using in situ Hi-C.[81] Low-C has been used to map the chromatin structure of primary B cells of a diffuse large B-cell lymphoma patient and was used to find high chromosome structural variation between the patient and healthy B-cells.
[23] Overall, the application of Hi-C and its variants in cancer research provides unique insight into the molecular underpinnings of the driving factors of cell abnormality.