Idiopathic pulmonary fibrosis
IPF should be considered in all patients with unexplained chronic exertional dyspnea who present with cough, inspiratory bilateral basal crackles, or finger clubbing.
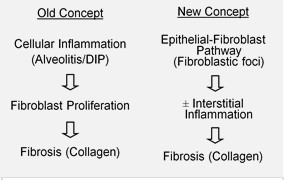
[3][17] IPF is believed to be the result of an aberrant wound healing process including/involving abnormal and excessive deposition of collagen (fibrosis) in the pulmonary interstitium with minimal associated inflammation.
Under pathologic conditions and in the presence of transforming growth factor beta (TGF-β), fibroblasts accumulate in these areas of damage and differentiate into myofibroblasts that secrete collagen and other proteins.
[25] This pathogenetic model is indirectly supported by the clinical features of IPF, including an insidious onset over several years, relatively infrequent acute exacerbations, and failure to respond to immunosuppressive therapy.
[27] A remarkable aspect of the MUC5B variant is its high frequency of detection, as it is found in approximately 20% of individuals with Northern and Western European ancestry and in 19% of the Framingham Heart Study population.
[3] However, in 2011, new simplified and updated criteria for the diagnosis and management of IPF were published by the ATS, ERS, together with the Japanese Respiratory Society (JRS) and Latin American Thoracic Association (ALAT).
The key issue facing clinicians is whether the presenting history, symptoms (or signs), radiology, and pulmonary function testing are collectively in keeping with the diagnosis of IPF or whether the findings are due to another process.
It has long been recognized that patients with ILD related to asbestos exposure, drugs (such as chemotherapeutic agents or nitrofurantoin), rheumatoid arthritis and scleroderma/systemic sclerosis may be difficult to distinguish from IPF.
IPF is one specific presentation of idiopathic interstitial pneumonia (IIP), which is in turn a type of ILD, also known as diffuse parenchymal lung disease (DPLD).
Examples of ILD of known cause include hypersensitivity pneumonitis, pulmonary Langerhan's cell histiocytosis, asbestosis, and collagen vascular disease.
[citation needed] Fibroblastic foci are dense collections of myofibroblasts and scar tissue and, together with honeycombing, are the main pathological findings that allow a diagnosis of UIP.
Pulmonary rehabilitation may alleviate the overt symptoms of IPF and improve functional status by stabilizing and/or reversing the extrapulmonary features of the disease.
[43] Typical programs of rehabilitation include exercise training, nutritional modulation, occupational therapy, education and psychosocial counseling.
In the first clinical trial of 180 patients (IFIGENIA), NAC was shown in previous study to reduce the decline in VC and DLCO over 12 months of follow-up when used in combination with prednisone and azathioprine (triple therapy).
[49] More recently, a large randomized, controlled trial (PANTHER-IPF) was undertaken by the National Institutes of Health (NIH) in the US to evaluate triple therapy and NAC monotherapy in IPF patients.
[42] Symptomatic patients with IPF younger than 65 years of age and with a body mass index (BMI) ≤26 kg/m2 should be referred for lung transplantation, but there are no clear data to guide the precise timing for LTx.
[62] a routine evaluation every 3 to 6 months, including spirometry (body plethysmography), diffusion capacity testing, chest X-rays, 6MWT, assessment of dyspnea, quality of life, oxygen requirement is mandatory.
[citation needed] In addition, the increasing awareness of complications and common concomitant conditions frequently associated with IPF requires a routinely evaluation of comorbidities, most of them simply reflecting concurrent diseases of aging, and medications with their interaction and side effects.
Acute exacerbations of IPF (AE-IPF) are defined as an unexplained worsening or development of dyspnea within 30 days with new radiological infiltrates at HRCT abnormality often superimposed on a background consistent with UIP pattern.
Many patients experiencing acute deterioration require intensive care treatment, particularly when respiratory failure is associated with hemodynamic instability, significant comorbidities or severe hypoxemia.
[67] The name of the index is GAP and is based on gender [G], age [A], and two lung physiology variables [P] (FVC and DLCO) that are commonly measured in clinical practice to predict mortality in IPF.
For example, it has been shown that IPF patients who have a specific genotype in the mucin MUC5B gene polymorphism (see above) experience slower decline in FVC and significantly improved survival.
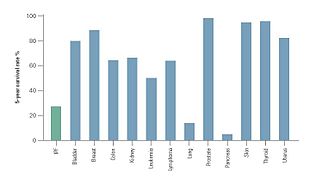
[10] The prevalence of IPF has been estimated between 14.0 and 42.7 per 100,000 persons based on a USA analysis of healthcare claims data, with variation depending on the case definitions used in this analyses.
In the 27 European Union countries, a range of sources estimate an incidence of 4.6–7.4 people per 100,000 of the population,[73][74] suggesting that approximately 30,000–35,000 new patients will be diagnosed with IPF each year.
IPF was the most common diagnosis (28%) followed by connective tissue disease-related ILD (14%), hypersensitivity pneumonitis (7%) and non-specific interstitial pneumonia (NSIP) (7%).
[76] Due to a heterogeneous distribution of the disease across European countries, epidemiological data needs to be updated through a Europe-wide registry for ILD and IPF.
A number of agents are currently being investigated in Phase II clinical trials for IPF, including the monoclonal antibodies simtuzumab, tralokinumab, lebrikizumab and FG-3019, a lysophosphatidic acid receptor antagonist (BMS-986020).
[80] These molecules are directed against several growth factors and cytokines that are known to play a role in the proliferation, activation, differentiation or inappropriate survival of fibroblasts.
[84] The algorithm outputs a score (ZCoR) using medical history on file with no new tests, and might be deployable as a universal IPF screening tool in primary care.
ZCoR has been trained and validated on nearly 3 million patients across multiple databases, achieving high predictive performance in out-of-sample data (positive likelihood ratio > 30 with 99% specificity).