Pulmonary hypertension
[7] Symptoms include shortness of breath, fainting, tiredness, chest pain, swelling of the legs, and a fast heartbeat.
[4] Risk factors include a family history, prior pulmonary embolism (blood clots in the lungs), HIV/AIDS, sickle cell disease, cocaine use, chronic obstructive pulmonary disease, sleep apnea, living at high altitudes, and problems with the mitral valve.
[4] Medications specifically used to treat pulmonary hypertension include epoprostenol, treprostinil, iloprost, bosentan, ambrisentan, macitentan, and sildenafil, tadalafil, selexipag, riociguat.
Signs of systemic congestion resulting from right-sided heart failure include jugular venous distension, ascites, and hepatojugular reflux.
[4] These guidelines are endorsed by the International Society for Heart and Lung Transplantation, and provide the current framework for understanding and treatment of pulmonary hypertension.
[25] The SMAD transcription factor family, including SMAD1, SMAD4, and SMAD9 are involved in signaling pathways downstream from BMPR2 and are also implicated in the development of pulmonary arterial hypertension.
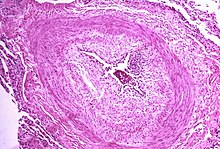
[25] The pathogenesis of pulmonary arterial hypertension (WHO Group I) involves the narrowing of blood vessels connected to and within the lungs.
The mechanisms involved in this narrowing process include vasoconstriction, thrombosis, and vascular remodeling (excessive cellular proliferation, fibrosis, and reduced apoptosis/programmed cell death in the vessel walls, caused by inflammation, disordered metabolism and dysregulation of certain growth factors).
This phenomenon is called hypoxic pulmonary vasoconstriction and it is initially a protective response to stop too much blood flowing to areas of the lung that are damaged and do not contain oxygen.
This combination of vessel occlusion and vascular remodeling once again increases the resistance to blood flow and so the pressure within the system rises.
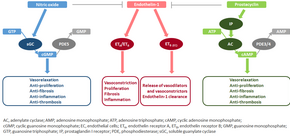
[39][40] The molecular mechanism of pulmonary arterial hypertension (PAH) is not known yet, but it is believed that the endothelial dysfunction results in a decrease in the synthesis of endothelium-derived vasodilators such as nitric oxide and prostacyclin.
[42] This nitric oxide diffuses into neighboring cells (including vascular smooth muscle cells and platelets), where it increases the activity of the enzyme soluble guanylate cyclase, leading to increased formation of cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP).
For example, the mitochondrial enzyme pyruvate dehydrogenase kinase (PDK) is pathologically activated in PAH, causing a metabolic shift from oxidative phosphorylation to glycolysis and leading to increased cell proliferation and impaired apoptosis.
[49] Other factors underlying the proliferative state of pulmonary vascular smooth muscle cells include OPG[52] and TRAIL.
[53] Focusing only on the pulmonary vasculature provides an incomplete picture of PAH; the ability of the right ventricle to adapt to the increased workload varies between patients and is an important determinant of survival.
[27] Even though the primary cause of PAH is unknown, inflammation and oxidative stress have been shown to have a key role in vascular remodeling.
[54] These factors are known to cause DNA damage, and may also promote the proliferative and apoptosis-resistant phenotype that is observed in PAH vascular cells.
[15] A physical examination is performed to look for typical signs of pulmonary hypertension (described above),[55] and a detailed family history is established to determine whether the disease might be heritable.
[60] Thus, Doppler echocardiography can suggest the presence of pulmonary hypertension, but right heart catheterization (described below) remains the gold standard for diagnosis of PAH.
[68] Patients with left heart failure or hypoxemic lung diseases (groups II or III pulmonary hypertension) should not routinely be treated with vasoactive agents including prostanoids, phosphodiesterase inhibitors, or endothelin antagonists, as these are approved for the different condition called primary pulmonary arterial hypertension.
Calcium channel blockers have been largely misused, being prescribed to many patients with non-vasoreactive PAH, leading to excess morbidity and mortality.
The trials supporting the use of these agents have been relatively small, and the only measure consistently used to compare their effectivity is the "six-minute walk test".
[73] Keros Therapeutics has decided to end a mid-stage trial of its experimental treatment, cibotercept, after identifying safety concerns, including fluid buildup around the heart in patients with PAH.
Three of these pathways are important since they have been targeted with drugs – endothelin receptor antagonists, phosphodiesterase type 5 (PDE-5) inhibitors, and prostacyclin derivatives.
[78] Moderate quality evidence suggests that endothelin receptor antagonists improve exercise capacity and decrease symptoms severity.
[81] A similar drug, ambrisentan (which is a ETA endothelin receptor blocker) is sold under the brand name Letairis in the US by Gilead Sciences.
It is the surgical removal of an organized thrombus (clot) along with the lining of the pulmonary artery; it is a very difficult, major procedure that is currently performed in a few select centers.
[94] Established clinical practice guidelines dictate the frequency of pulmonary nodule evaluation and surveillance,[69][95] patients are normally monitored through commonly available tests such as:[citation needed] PAH is considered a universally fatal illness, although survival time may vary between individuals.
The prognosis of pulmonary arterial hypertension (WHO Group I) has an untreated median survival of 2–3 years from time of diagnosis, with the cause of death usually being right ventricular failure (cor pulmonale).
[40] A small percentage of patients with COPD develop pulmonary hypertension with no other disease to explain the high pressure.