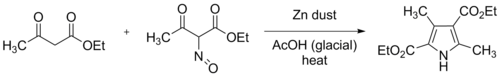Knorr pyrrole synthesis
[5][6] The original Knorr synthesis employed two equivalents of ethyl acetoacetate, one of which was converted to ethyl 2-oximinoacetoacetate by dissolving it in glacial acetic acid, and slowly adding one equivalent of saturated aqueous sodium nitrite, under external cooling.
The reaction is exothermic, and the mixture can reach the boiling point, if external cooling is not applied.
Dissolving Knorr's pyrrole in concentrated sulfuric acid, and then pouring the resulting solution into water will hydrolyze the 4-ester group selectively.
The mechanism of the Knorr pyrrole synthesis begins with condensation of the amine and ketone to give an imine.
[12] George Kleinspehn reported that the Fischer–Fink connectivity could be forced to occur exclusively, by the use of diethyl oximinomalonate in the synthesis, with 2,4-pentanedione, or its 3-alkyl substituted derivatives.
[14] Meanwhile, Johnson had extended the Fischer-Fink synthesis by reacting 2-oximinoacetoacetate esters (ethyl, benzyl, or tertiary-butyl), with 3-alkyl substituted 2,4-pentanediones.
[15] The Kleinspehn synthesis was extended under David Dolphin by the use of unsymmetrical β-diketones (such as 3-alkyl substituted 2,4-hexanediones), which preferentially reacted initially at the less hindered acetyl group and afforded the corresponding 5-methylpyrrole-2-carboxylate esters.
N,N-Dialkyl 2-oximinoacetoacetamides also were found to give pyrroles when reacted under Knorr conditions with 3-substituted-2,4-pentanediones, in yields comparable to the corresponding esters (around 45%).
[16] This same mechanism occurs to a minor extent in the acetoacetate ester systems, and had previously been detected radiochemically by Harbuck and Rapoport.