Multi-state modeling of biomolecules
Multi-state modeling of biomolecules refers to a series of techniques used to represent and compute the behaviour of biological molecules or complexes that can adopt a large number of possible functional states.
Development of faster and more powerful methods is ongoing, promising the ability to simulate ever more complex signaling processes in the future.
[22] These networks rely on the ability of single proteins to exist in a variety of functionally different states achieved through multiple mechanisms, including post-translational modifications, ligand binding, conformational change, or formation of new complexes.
In addition, several types of modifications can co-exist, exerting a combined influence on a biological macromolecule at any given time.
Biological signaling networks incorporate a wide array of reversible interactions, post-translational modifications and conformational changes.
A model of coupling of the EGF receptor to a MAP kinase cascade presented by Danos and colleagues[29] accounts for
The existence (or potential existence) of such large numbers of molecular species is a combinatorial phenomenon: It arises from a relatively small set of features or modifications (such as post-translational modification or complex formation) that combine to dictate the state of the entire molecule or complex, in the same way that the existence of just a few choices in a coffee shop (small, medium or large, with or without milk, decaf or not, extra shot of espresso) quickly leads to a large number of possible beverages (24 in this case; each additional binary choice will double that number).
(In the coffee shop example, adding an extra shot of espresso will cost 40 cent, no matter what size the beverage is and whether or not it has milk in it).
This means that even a model that can account for a vast number of molecular species and reactions is not necessarily conceptually complex.
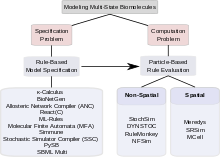
The combinatorial complexity of signaling systems involving multi-state proteins poses two kinds of problems.
Among the approaches that have been proposed to tackle combinatorial complexity in multi-state modeling, some are mainly concerned with addressing the specification problem, some are focused on finding effective methods of computation.
An early rule-based specification method is the κ-calculus,[1] a process algebra that can be used to encode macromolecules with internal states and binding sites and to specify rules by which they interact.
[6] Conceptually, ANC sees molecules as allosteric devices with a Monod-Wyman-Changeux (MWC) type regulation mechanism,[36] whose interactions are governed by their internal state, as well as by external modifications.
[42] A similar nested language to represent multi-level biological systems has been proposed by Oury and Plotkin.
For instance, the Simmune project[44][45] includes a spatial component: Users can specify their multi-state biomolecules and interactions within membranes or compartments of arbitrary shape.
The Stochastic Simulator Compiler (SSC)[46] allows for rule-based, modular specification of interacting biomolecules in regions of arbitrarily complex geometries.
This allows users to store higher-order biochemical processes such as catalysis or polymerisation as macros and re-use them as needed.
[34] Models involving multi-state and multi-component species can also be specified in Level 3 of the Systems Biology Markup Language (SBML) [34] using the multi package.
One way to classify simulation algorithms is by looking at the level of analysis at which the rules are applied: they can be population-based, single-particle-based or hybrid.
[3][5] Even if a large reaction system can be successfully generated, its simulation using population-based rule evaluation can run into computational limits.
[51] In cases of combinatorial complexity, however, the modeling of individual particles is an advantage because, at any given point in the simulation, only existing molecules, their states and the reactions they can undergo need to be considered.
This method reduces the complexity of the model at the simulation stage, and thereby saves time and computational power.
In StochSim, each molecular species can be equipped with a number of binary state flags representing a particular modification.
These properties make StochSim ideally suited to modeling multi-state molecules arranged in holoenzymes or complexes of specified size.
The Network-Free Stochastic Simulator (NFSim) differs from those described above by allowing for the definition of reaction rates as arbitrary mathematical or conditional expressions and thereby facilitates selective coarse-graining of models.
[53] It is easy to imagine a biological system where some components are complex multi-state molecules, whereas others have few possible states (or even just one) and exist in large numbers.
[56] MCell[19][20][21] allows individual molecules to be traced in arbitrarily complex geometric environments which are defined by the user.
This can in some cases be circumvented by adjusting the diffusion constants of ligands that interact with the complex, by using checkpointing functions or by combining simulations at different levels.
A (by no means exhaustive) selection of models of biological systems involving multi-state molecules and using some of the tools discussed here is give in the table below.
This article was adapted from the following source under a CC BY 4.0 license (2014) (reviewer reports): Melanie I Stefan; Thomas M Bartol; Terrence J Sejnowski; Mary B Kennedy (September 2014).