Noonan syndrome
Noonan syndrome (NS) is a genetic disorder that may present with mildly unusual facial features, short height, congenital heart disease, bleeding problems, and skeletal malformations.
[3][1] Noonan syndrome is a type of RASopathy, the underlying mechanism for which involves attenuation of the RAS/MAPK cell signaling pathway.
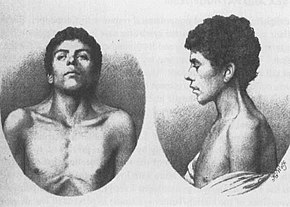
[2] The most common signs leading to the diagnosis of Noonan syndrome are unique facial characteristics and musculoskeletal features.
For short stature, growth hormone is sometimes combined with IGF-1 (or as an alternative, IGF-1 as a stand-alone) can be used to achieve an increased height/final height quicker.
50-70% of individuals with NS are born with some form of congenital heart defect, with pulmonary valvular stenosis being the most common (50–60%).
These include swallowing difficulties, low gut motility, gastroparesis (delayed gastric emptying), intestinal malrotation, and frequent or forceful vomiting.
These digestive issues may lead to decreased appetite, failure to thrive from infancy to puberty (75%), and occasionally the need for a feeding tube.
Lymphatic anomalies including Posterior cervical hygroma (webbed neck) and Lymphedema may present in people with Noonan syndrome.
It has been associated with Von Willebrand disease, Amegakaryocytic thrombocytopenia (low platelet count), prolonged activated partial thromboplastin time, combined coagulation defects.
[17] Individuals may experience bleeding disorders of various types, often associated with thrombocytopenia, low levels of clotting factors, impaired platelet function, and more.
[18][19] Recurrence in siblings and apparent transmission from parent to child has long suggested a genetic defect with autosomal dominant inheritance and variable expression.
However, while 30-75% of cases show a noticeable inheritance from one of the parents, the rest are caused by de-novo genetic mutations occurring for the first time in the affected individual.
[23][24][25] Heterozygous mutations in NRAS, HRAS, BRAF, SHOC2, MAP2K1, MAP2K2, and CBL have also been associated with a smaller percentage of NS and related phenotypes.
The principal values of making a genetic diagnosis are that it guides additional medical and developmental evaluations, it excludes other possible explanations for the features, and it allows more accurate recurrence risk estimates.
With more genotype-phenotype correlation studies being performed, a positive genetic diagnosis will help the clinician to be aware of possible anomalies specific to that certain gene mutation.
In the future, studies may lead to a targeted management of NS symptoms that depends on what genetic mutation a person has.
Prenatal features that might lead physicians to consider a diagnosis of Noonan syndrome include cystic hygroma, increased nuchal translucency, pleural effusion, and edema.
[45][8] Jacqueline Noonan began practicing as a pediatric cardiologist in 1959 at the University of Iowa when she noticed that children with a rare type of heart defect, valvular pulmonary stenosis, often had a characteristic physical appearance, with short stature, webbed neck, wide spaced eyes, and low-set ears.
[46] This described nine children who in addition to congenital heart disease had characteristic facial features, chest deformities and short stature.