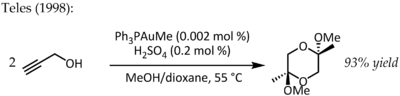Organogold chemistry
These gold sources, however, quickly give rise to ill-defined and easily deactivated (via reduction to Au0) active catalysts in solution.
[10][11] Although the coordinatively unsaturated complex "LAu+" is notionally generated from a LAuCl/AgX mixture, the exact nature of the cationic gold species and the role of the silver salt remains somewhat contentious.
Electrophilic ions and complexes such as these with a strong propensity to form π-complexes are generally known as pi(π)-acids (see also: cation–pi interaction).
In oxymercuration the resultant organomercurial species is generated stoichiometrically, and requires an additional step to liberate the product.
In the case of gold, protonolysis of the Au-C bond closes the catalytic cycle, allowing the coordination of another substrate.
Some practical advantages of gold(I) catalysis include: 1) air stability (due to the high oxidation potential of Au(I)), 2) tolerance towards adventitious moisture (due its low oxophilicity), and 3) relatively low toxicity compared to other pi-acids (e.g., Pt(II) and Hg(II)).
[18] Teles identified a major drawback of this method as Au(III) was rapidly reduced to catalytically dead metallic gold and in 1998 returned to the theme of ligand supported Au(I) for the same transformation:[19] This particular reaction demonstrated fantastic catalytic efficiency and would trigger a flurry of research into the use of phosphinegold(I) complexes for the activation C-C multiple bonds in the years to come.
[20] In spite of the lower stability of gold(III) complexes under catalytic conditions, simple AuCl3 was also found to be an efficient catalyst in some cases.
For instance, Hashmi reported an AuCl3-catalyzed alkyne / furan Diels–Alder reaction - a type of cycloaddition that does not ordinarily occur - for the synthesis of 2,3-disubstituted phenols:[21] Further mechanistic studies conclude that this is not a concerted transformation, but rather an initial alkyne hydroarylation followed by a series of non-obvious intramolecular rearrangements, concluding with a 6π electrocyclization and rearomatization.
Relativistic effects are significant in organogold chemistry due to the large nuclear charge of the metal (Z = 79).
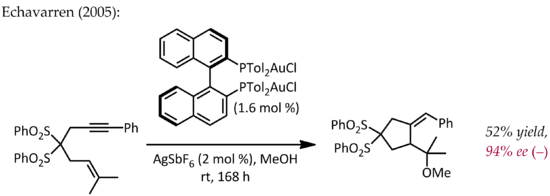
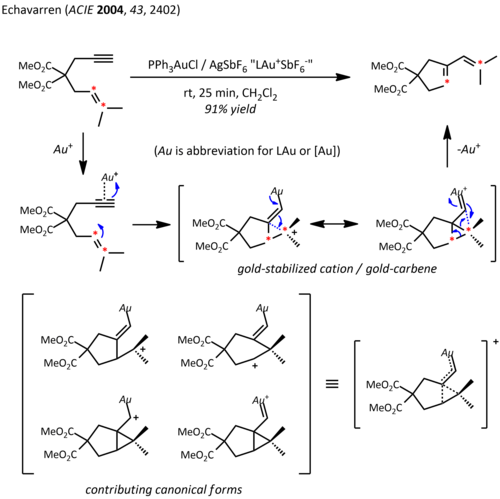
Thus, in addition to their expected carbocation-like reactivity, these cations also exhibit significant carbene character, a property that has been exploited in catalytic transformations such as cyclopropanation and C-H insertion.
[22] Propargyl esters can serve as precursors for cationic gold-vinylcarbene intermediates, which can react with alkenes in a concerted manner to afford the cyclopropanation product.
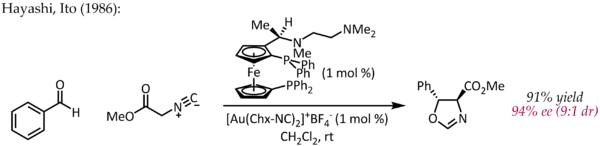
During the past decade, several studies have demonstrated that gold can efficiently catalyze C-C and C-heteroatom cross-coupling reactions that proceed through an Au(I)/Au(III) cycle.
[34] While gold-catalyzed hydrofunctionalization of alkynes, allenes, and allylic alcohols[35] occurs readily under comparatively mild conditions, unactivated alkene remain poor substrates in most cases,[36] in large part due to the resistance of the intermediate alkylgold(I) complexes to protodeauration.