Heterogeneous gold catalysis
[1] Besides enabling an optimal dispersion of the nanoclusters, the support materials have been suggested to promote catalysis by altering the size, shape, strain and charge state of the cluster.
[3][7][8] A precise shape control of the deposited gold clusters has been shown to be important for optimizing the catalytic activity, with hemispherical, few atomic layers thick nanoparticles generally exhibiting the most desirable catalytic properties due to maximized number of high-energy edge and corner sites.
Very recently, there has been a lot of developments in gold catalysis for the synthesis of organic molecules including the C-C bond forming homocoupling or cross-coupling reactions and it has been speculated that some of these catalysts could find applications in various fields.
The turnover frequency (TOF) of CO oxidation on these cationic gold catalysts is in the order of magnitude of 0.01 s−1, exhibiting the very high activation energy of 138 kJ/mol.
[2] Supported gold nanoclusters with a diameter < 2 nm are active to CO oxidation with turnover number (TOF) in the order of magnitude of 0.1 s−1.
Metal hydroxide supports such as Be(OH)2, Mg(OH)2, and La(OH)3, with gold clusters of < 1.5 nm in diameter constitute highly active catalysts for CO oxidation at 200 K (-73 °C).
By means of techniques such as HR-TEM and EXAFS, it has been proven that the activity of these catalysts is due exclusively to clusters with 13 atoms arranged in an icosahedron structure.
In the case of powder catalysts prepared by wet methods, the surface OH− groups on the support provide sufficient aid as co-catalysts, so that no additional moisture is necessary.
According to DFT calculations, the presence of such Au cations on the catalyst is allowed by empty, localized nonbonding f states in CeO2.
The active species in these catalysts were identified to be hemispherical gold nano-crystals of less than 2 nm in diameter in intimate contact with the support.
[2] Notwithstanding its poor activity, nano-sized gold immobilized in various supports has been found to provide a good selectivity in hydrogenation reactions.
In later works, it was shown that gold-catalyzed hydrogenation can be highly sensitive to Au loading (hence to particle size) and to the nature of the support.
[12] By contrast, the hydrogenation of 1,3-butadiene to 1-butene was shown to be relatively insensitive to Au particle size in a study with a series of Au/Al2O3 catalysts prepared by different methods.
Because the reaction with deuterium was substantially slower, it was suggested that the rate-determining step in alkene hydrogenation was the cleavage of the H-H bond.
Gold catalysts are able to hydrogenate only the carbonyl group, so that the aldehyde is transformed to the corresponding alcohol, while leaving the C=C double bond untouched.
[12] A strategy that in many reactions has succeeded at improving gold's catalytic activity without impairing its selectivity is to synthesize bimetallic Pd-Au or Pt-Au catalysts.
[14][15] Bulk metallic gold is known to be inert, exhibiting a surface reactivity at room temperature only towards a few substances such as formic acid and sulphur-containing compounds, e.g. H2S and thiols.
However, if the adsorption is weak such as in the case of bulk gold, a sufficient perturbation of the reactant electronic structure does not occur and catalysis is hindered (Sabatier's principle).
When gold is deposited as nanosized clusters of less than 5 nm onto metal oxide supports, a markedly increased interaction with adsorbates is observed, thereby resulting in surprising catalytic activities.
[3][13][16] It is generally known that decreasing the size of metallic particles in some dimension to the nanometer scale will yield clusters with a significantly more discrete electronic band structure in comparison with the bulk material.
[9] This is an example of a quantum-size effect and has been previously correlated with an increased reactivity enabling nanoparticles to bind gas phase molecules more strongly.
Nevertheless, the observed trends provide further evidence that a significant perturbation of the Au electronic structure occurs upon nanoscaling, which is likely to play a key role in the enhancement of the catalytic properties of gold nanoparticles.
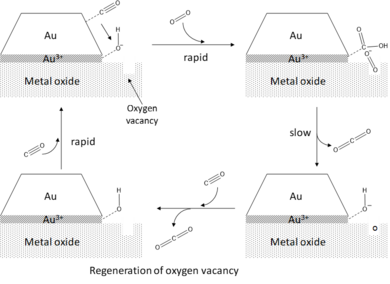
[1][2] In the case of CO oxidation, it has been hypothesized that CO adsorbs onto the edges and corners of the gold clusters, while the activation of oxygen occurs at the peripheral sites.
The low degree of coordination increases the surface energy of corner and edge sites, hence making them more active towards binding adsorbates.
[8] This charge transfer induces a local perturbation in the electronic structure of the gold clusters at the perimeter sites, enabling the formation of resonance states as the antibonding
In gas-phase model studies, the formation of activated super-oxo species O2− is found to correlate with the size-dependent electronic properties of the clusters.
[21][22] The activation of O2 at the perimeter sites is also observed for defect-free surfaces and neutral gold clusters, but to a significantly smaller extent.
[9] The induced strains especially affect the Au atoms close to the substrate-cluster interface, resulting in a shift of the local d-band center towards energies closer to the Fermi level.
This illustrates the ability to fine-tune the catalytic activity of gold clusters via varying the support material as well as the underlying metal upon which the substrate has been grown.
[2] Invoking the periphery hypothesis, water promotes the activation of O2 by co-adsorption onto the perimeter sites where it reacts with O2 to form adsorbed hydroxyl (OH*) and hydroperoxo (OOH*) species.