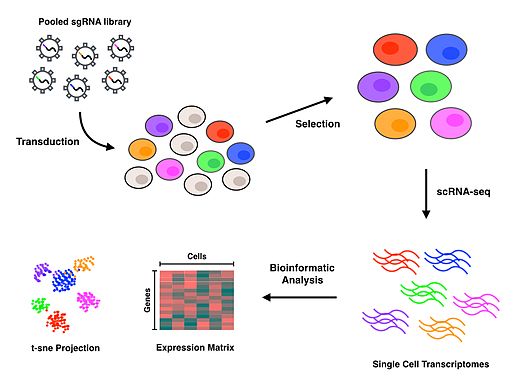
Perturb-seq
Perturb-seq (also known as CRISP-seq and CROP-seq) refers to a high-throughput method of performing single cell RNA sequencing (scRNA-seq) on pooled genetic perturbation screens.
Perturb-seq is a reverse genetics approach that allows for the investigation of phenotypes at the level of the transcriptome, to elucidate gene functions in many cells, in a massively parallel fashion.
Upon completion of the protocol, bioinformatics analyses are conducted to associate each specific cell and perturbation with a transcriptomic profile that characterizes the consequences of inactivating each gene.
[3] While each paper shared the core principles of combining CRISPR mediated perturbation with scRNA-seq, their experimental, technological and analytical approaches differed in several aspects, to explore distinct biological questions, demonstrating the broad utility of this methodology.
Knockout libraries perturb genes through double stranded breaks that prompt the error prone non-homologous end joining repair pathway to introduce disruptive insertions or deletions.
CRISPR interference (CRISPRi) on the other hand utilizes a catalytically inactive nuclease to physically block RNA polymerase, effectively preventing or halting transcription.
[1][4][11] The authors who first performed Perturb-seq developed an in-house computational framework called MIMOSCA that predicts the effects of each perturbation using a linear model and is available on an open software repository.
[12] Perturb-seq makes use of current technologies in molecular biology to integrate a multi-step workflow that couples high-throughput screening with complex phenotypic outputs.
The preparation of these extensive libraries depends upon a comparative increase in the resources required to culture the massive numbers of cells that are needed to achieve a successful screen of many perturbations.
Recently, the Perturb-seq (CROP-seq) workflow has been adapted to enable genome-scale CRISPRi (CRISPR interference) screens in Jurkat cells at single-cell resolution.
In total, one million Jurkat cells were processed for single-cell RNA sequencing allowing transcriptomic readouts of a final list of 374 marker genes involved in TCR signaling.
The bioinformatic analysis confirmed more than 70 known activators and repressors of TCR signaling cascades, hence showcasing the potential of Perturb-seq (CROP-seq) screens to support translational research.