Phenylalanine hydroxylase
PAH is one of three members of the biopterin-dependent aromatic amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin (BH4, a pteridine cofactor) and a non-heme iron for catalysis.
Two pathways have been proposed based on models that differ in the proximity of the iron to the pterin cofactor and the number of water molecules assumed to be iron-coordinated during catalysis.
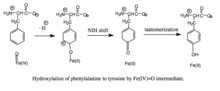
The activated O2 then attacks BH4, forming a transition state characterized by charge separation between the electron-deficient pterin ring and the electron-rich dioxygen species.
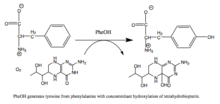
[7] When productive, though, the Fe(IV)=O intermediate is added to phenylalanine in an electrophilic aromatic substitution reaction that reduces iron from the ferryl to the ferrous state.
[15] Many studies suggest that mammalian PAH shows behavior comparable to porphobilinogen synthase (PBGS), wherein a variety of factors such as pH and ligand binding are reported to affect enzyme activity and protein stability.
[20] Inclusion of a Phe analogue in the crystal structure changes both iron from a six- to a five-coordinated state involving a single water molecule and bidentate coordination to Glu330 and opening a site for oxygen to bind.
[23] Hydrogen/deuterium exchanges analysis indicates that allosteric binding of Phe globally alters the conformation of PAH such that the active site is less occluded as the interface between the regulatory and catalytic domains is increasingly exposed to solvent.
Deletion of the N-terminal domain also eliminates the lag time while increasing the affinity for Phe by nearly two-fold; no difference is observed in the Vmax or Km for the tetrahydrobiopterin cofactor.
[26] Additional regulation is provided by Ser16; phosphorylation of this residue does not alter enzyme conformation but does reduce the concentration of Phe required for allosteric activation.
The resulting distortion of the tetramer symmetry is evident in the differential surface area of the dimerization interfaces and distinguishes PAH from the tetramerically symmetrical tyrosine hydroxylase.
[27] PAH is a critical enzyme in phenylalanine metabolism and catalyzes the rate-limiting step in its complete catabolism to carbon dioxide and water.
[13][28] Regulation of flux through phenylalanine-associated pathways is critical in mammalian metabolism, as evidenced by the toxicity of high plasma levels of this amino acid observed in phenylketonuria (see below).
[28] PKU is both genotypically and phenotypically heterogeneous: Over 300 distinct pathogenic variants have been identified, the majority of which correspond to missense mutations that map to the catalytic domain.
[13] BH44 has been administered as a pharmacological treatment and has been shown to reduce blood levels of phenylalanine for a segment of PKU patients whose genotypes lead to some residual PAH activity but have no defect in BH44 synthesis or regeneration.
Now, PKU is part of newborn screening in many countries, and elevated phenylalanine levels are identified shortly after birth by measurement with tandem mass spectrometry.