Pituitary adenoma
[1] Pituitary adenomas represent from 10% to 25% of all intracranial neoplasms, with an estimated prevalence rate in the general population of approximately 17%.
[2][4] The majority of pituitary microadenomas remain undiagnosed, and those that are diagnosed are often found as an incidental finding and are referred to as incidentalomas.
Some tumors secrete more than one hormone, the most common combination[8] being GH and prolactin, which present as gigantism or acromegaly and unexpected lactation (in both men and women).
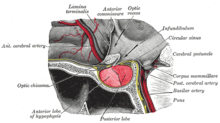
[citation needed] A patient with pituitary adenoma may present with visual field defects, classically on the left and right in bitemporal hemianopsia.
If originating superior to the optic chiasm, more commonly in a craniopharyngioma of the pituitary stalk, the visual field defect will first appear as bitemporal inferior quadrantanopia.
[15] Non-secreting adenomas can go undetected for an extended time because no obvious abnormalities are seen; the gradual reduction in normal activities due to decreased production of hormones is rather less evident.
For example, insufficient adrenocorticotropic hormone means that the adrenal glands will not produce sufficient cortisol, resulting in slow recovery from illness, inflammation, and chronic fatigue; insufficient growth hormone in children and adolescents leads to diminished stature but which can have many other explanations.
Psychiatric symptoms such as depression, anxiety[16] apathy, emotional instability, easy irritability and hostility have been noted.
In some patients with CNC, the pituitary gland is characterized by hyperplastic areas with the hyperplasia most likely preceding the formation of GH-producing adenomas.
[31] Familial isolated pituitary adenoma (FIPA) is a term that is used to identify a condition that displays an autosomal dominant inheritance and is characterised by the presence of two or more related patients affected by adenomas of the pituitary gland only, with no other associated symptoms that occur in multiple endocrine neoplasia type 1 (MEN-1), Carney complex and with mutations in the aryl hydrocarbon receptor-interacting protein (AIP) gene.
[39] Due to their young age at onset, AIP mutations are the most frequent genetic cause of pituitary gigantism (29% of cases).
[41][38] The disease characteristics of very young onset pituitary gigantism leads to severe overgrowth if not treated adequately; many of the tallest humans in history (e.g. Robert Pershing Wadlow; Sandy Allen, André Rousimoff (Andre the Giant), Zeng Jinlian) had a similar clinical history to patients with X-LAG syndrome.
Part of the hypothalamic-pituitary axis, it controls most of the body's endocrine functions via the secretion of various hormones into the circulatory system.
[48] The diagnosis is confirmed by testing hormone levels, and by radiographic imaging of the pituitary (for example, by CT scan or MRI).
[2] While non-secreting, noninvasive pituitary microadenomas are generally considered to be literally as well as clinically benign, a 2011 meta-analysis of available research showed there were, to that time, scant studies of low quality to support this assertion.
[3] The Clinical Practice Guidelines, as published in April 2011 in The Journal of Clinical Endocrinology and Metabolism by the Endocrine Society (a professional, international medical organization), recommend that all patients with pituitary incidentalomas undergo a complete medical history and physical examination, laboratory evaluations to screen for hormone hypersecretion and for hypopituitarism.
Other commonly reported symptoms include anterior pituitary dysfunction, visual field defects, headache/pain, and ophthalmoplegia.