Protecting group
[1] In many preparations of delicate organic compounds, specific parts of the molecules cannot survive the required reagents or chemical environments.
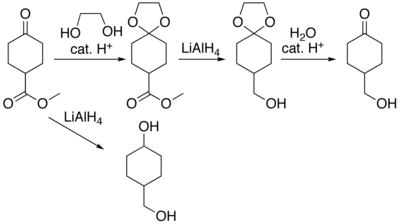
For example, lithium aluminium hydride is a highly reactive reagent that usefully reduces esters to alcohols.
Protecting groups are more common in small-scale laboratory work and initial development than in industrial production because they add additional steps and material costs.
Also, cheap chiral protecting groups may often shorten an enantioselective synthesis (e.g. shikimic acid for oseltamivir).
Apparent problems in synthesis strategies with protecting groups are rarely documented in the academic literature.
Each type of counterion, i.e. cleavage reagent, can also selectively cleave different silicon protecting groups depending on steric hindrance.
Catalytic hydrogenation removes a wide variety of benzyl groups: ethers, esters, urethanes, carbonates, etc.
Sterically hindered esters are less susceptible to nucleophilic attack: Triorganosilyl sources have quite variable prices, and the most economical is chlorotrimethylsilane (TMS-Cl), a Direct Process byproduct.
The trimethylsilyl ethers are also extremely sensitive to acid hydrolysis (for example silica gel suffices as a proton donator) and are consequently rarely used nowadays as protecting groups.
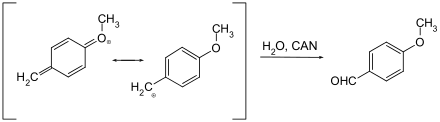
Aliphatic methyl ethers cleave with difficulty and only under drastic conditions, so that these are in general only used with quinonic phenols.
[45][46] Amines have a special importance in peptide synthesis, but are a quite potent nucleophile and also relatively strong bases.
Other, more exotic amine protectors are the phthalimides, which admit reductive cleavage,[48] and the trifluoroacetamides, which hydrolyze easily in base.
Indoles, pyrroles und imidazoles — verily any aza-heterocycle — admit protection as N‑sulfonylamides,which are far too stable with aliphatic amines.
[49] N‑benzylated amines can be removed through catalytic hydrogenation or Birch reduction, but have a decided drawback relative to the carbamates or amides: they retain a basic nitrogen.
Consequently acyclic acetals are used practically only when a very mild cleavage is required or when two different protected carbonyl groups must be differentiated in their liberation.
Thiols, which one begins with to form these acetals, have a very unpleasant stench and are poisonous, which severely limit their applications.
In contradistinction to the O,O‑acetal case, it is not needed to remove water from the reaction mixture in order to shift the equilibrium.
[69] Many groups can suffice for the alcoholic component, and the specific cleaving conditions are contrariwise generally quite similar: each ester can be hydrolyzed in a basic water-alcohol solution.
This normally proceeds from deprotonation (via a strong base like methylmagnesium bromide or butyllithium in tetrahydrofuran/dimethylsulfoxide) and subsequently reaction with chlorotrimethylsilane to a terminally TMS-protected alkyne.
[96] In order to protect the triple bond itself, sometimes a transition metal-alkyne complex with dicobalt octacarbonyl is used.
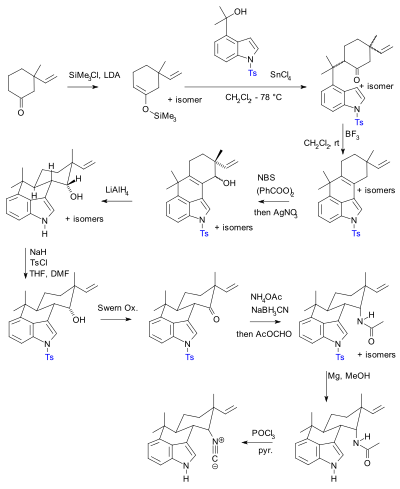
[103] In practical terms their use adds two steps (protection-deprotection sequence) to a synthesis, either or both of which can dramatically lower chemical yield.
The previously published synthesis[104][105][106] according to Baran, contained 20 steps with multiple protective group manipulations (two confirmed): Although the use of protecting groups is not preferred in industrial syntheses, they are still used in industrial contexts, e.g. sucralose (sweetener) or the Roche synthesis of oseltamivir (Tamiflu, an antiviral drug) An important example of industrial applications of protecting group theory is the synthesis of ascorbic acid (Vitamin C) à la Reichstein.
A tricoordinate phosphorus, used on account of the high reactivity, is tagged with a cyanoethyl protecting group on a free oxygen.